
Maison >Périphériques technologiques >IA >Une équipe chinoise a développé avec succès l'IA pour prédire les médicaments appropriés pour les patients atteints de cancer, et les résultats ont été publiés dans la revue Nature.
Une équipe chinoise a développé avec succès l'IA pour prédire les médicaments appropriés pour les patients atteints de cancer, et les résultats ont été publiés dans la revue Nature.

- PHPzavant
- 2023-04-20 21:43:121306parcourir
Avec une seule IA, les réponses cliniques de 9 808 patients atteints de cancer aux médicaments peuvent être entièrement prédites.
Et les résultats sont cohérents avec les observations cliniques.
Il s'agit du dernier résultat CODE-AE (autoencodeur déconfondant sensible au contexte) apporté par l'équipe de Lei Xie à la City University de New York.

Il propose un nouveau modèle d'auto-encodage contextuel capable de prédire les réponses spécifiques de différents patients aux médicaments.
Cela aura un impact significatif sur le développement de nouveaux médicaments et les essais cliniques.
Vous devez savoir que dans le modèle traditionnel, il faut près de 10 ans pour qu'un nouveau médicament soit développé, testé et entièrement lancé, et les fonds consommés sont d'une ampleur sans précédent, atteignant facilement 1 milliard de dollars américains.
Le cycle est si long parce que la réaction des nouveaux médicaments dans le corps humain est difficile à prédire et des essais répétés sont souvent nécessaires pour les tests.
Et si l’IA peut utiliser les données pour faire des prédictions, elle réduira considérablement les délais de commercialisation des nouveaux médicaments et réduira les coûts.
Actuellement, cette recherche a été publiée dans la sous-revue Nature "Nature Machine Intelligence".
En termes simples, CODE-AE utilise les données issues de la validation cellulaire in vitro de nouveaux médicaments pour prédire la réponse du médicament dans le corps humain.
Cela évite la dépendance de la formation des modèles d'IA aux données cliniques des patients.
La principale raison pour laquelle l'IA n'a pas été très efficace dans la prédiction de la réponse clinique dans le passé est qu'il est trop difficile de collecter des données massives et continues sur la réponse clinique.
Du point de vue du mécanisme, les chercheurs divisent les biomarqueurs des médicaments en domaines source et domaine cible.
Le domaine source représente un domaine différent de l'échantillon de test, mais contient de riches informations de supervision, qui peuvent être comprises comme des données de vérification cellulaire in vitro.
Le domaine cible est le domaine où se trouve l'échantillon de test. Il n'a pas d'étiquettes ou seulement quelques étiquettes, c'est-à-dire les données du patient.
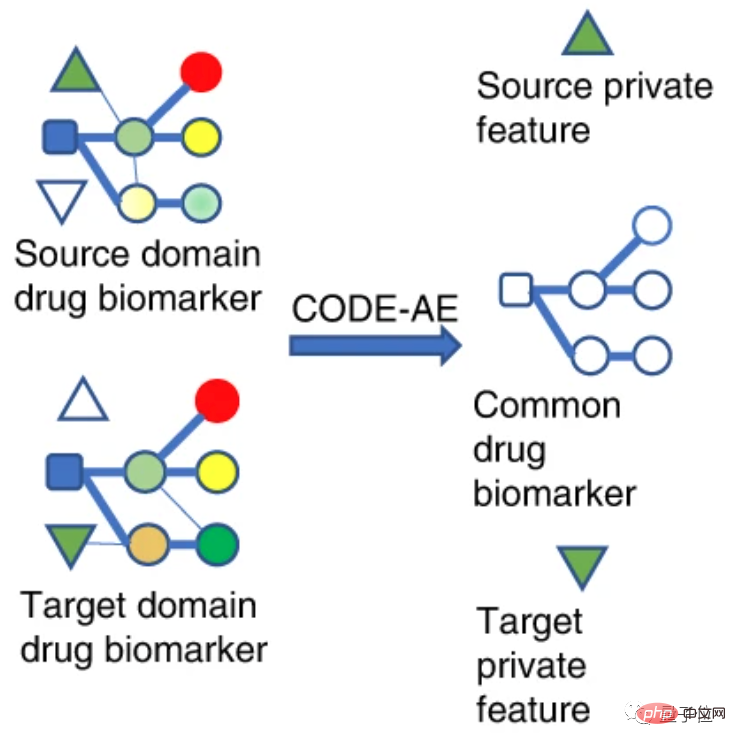
Mappez les caractéristiques de données de différents champs dans le même espace de caractéristiques afin que leurs distances dans cet espace soient aussi proches que possible.
Ainsi, la fonction objectif formée sur le domaine source dans l'espace des fonctionnalités peut être transférée vers le domaine cible pour améliorer la précision dans le domaine cible.
Dans le contexte de cette recherche, le domaine source et le domaine cible sont tous deux des caractéristiques de données des biomarqueurs de médicaments, c'est-à-dire des caractéristiques de données de cibles de médicaments.
En ce qui concerne spécifiquement le cadre du modèle, il est principalement divisé en trois parties : pré-entraînement, réglage fin et inférence.
La pré-formation utilise principalement l'apprentissage auto-supervisé pour créer un module de codage de fonctionnalités permettant de cartographier les profils d'expression génique non étiquetés des données cellulaires in vitro et des données des patients dans l'espace d'intégration. De cette façon, certains facteurs de confusion peuvent être éliminés et la distribution latente des deux données peut être cohérente pour éliminer les biais systématiques.
L'étape de mise au point consiste à ajouter un modèle supervisé sur la base d'un pré-entraînement et à utiliser des données cellulaires in vitro étiquetées pour l'entraînement.
Enfin, lors de la phase d'inférence, les patients obtenus lors de la pré-formation sont d'abord levés et intégrés, puis le modèle optimisé est utilisé pour prédire la réponse du patient au médicament.

Dans ce mode, CODE-AE a deux fonctionnalités.
Premièrement, il peut extraire des signaux biologiques communs et des représentations privées dans des échantillons incohérents, éliminant ainsi les interférences causées par différents modèles de données.
Deuxièmement, après avoir séparé le signal de réponse au médicament et les facteurs de confusion, un alignement local peut également être obtenu.
En résumé, CODE-AE peut être compris comme le processus de sélection de caractéristiques uniques dans le modèle de données incohérent intégrant l'espace de données étiquetées et non étiquetées.
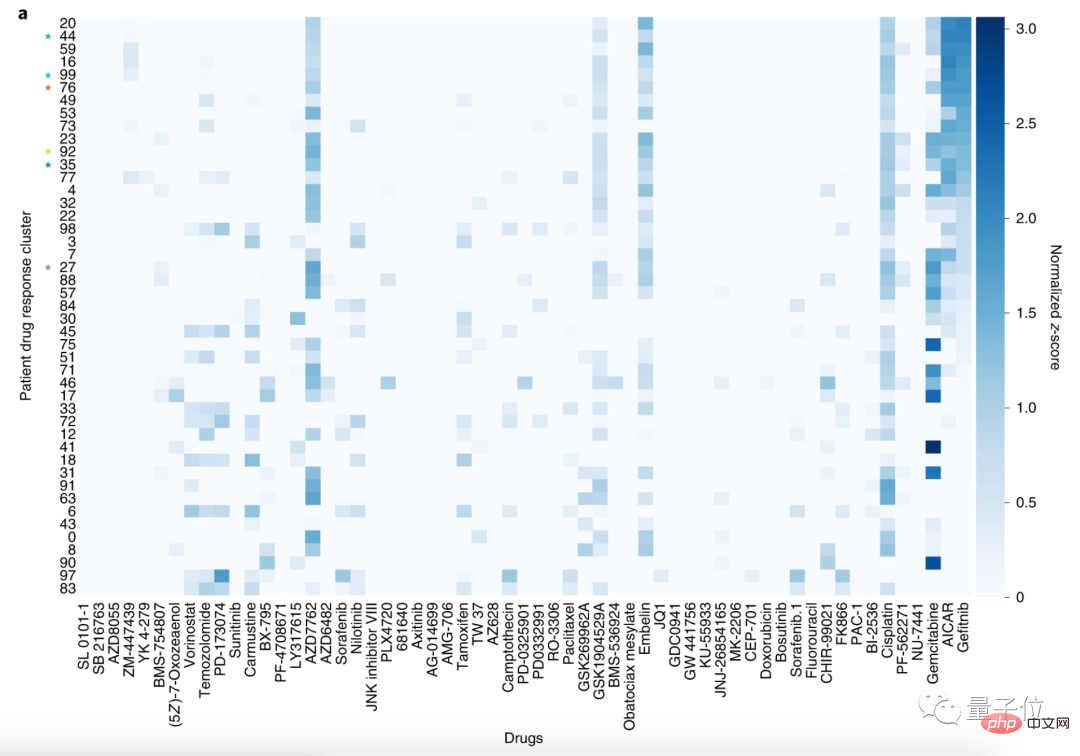
Pour démontrer l'efficacité du modèle, les chercheurs ont prédit l'adéquation du médicament à 9 808 patients atteints de cancer.
Si les résultats de site prédits par le modèle pour l'état du patient sont liés à la cible médicamenteuse qu'il utilise, cela prouve que la prédiction est correcte.
Les chercheurs ont ensuite divisé les patients en 100 clusters et les 59 médicaments en 30 clusters.
Grâce à cette méthode d'analyse, les patients présentant des profils de réponse médicamenteuse similaires peuvent être regroupés.
Ici, nous prenons comme exemple le regroupement de patients atteints d'un carcinome épidermoïde du poumon (LSCC) et d'un cancer du poumon non à petites cellules (NSCLC).
Parmi les 59 médicaments, les médicaments les plus sensibles au LSCC sont le géfitinib, l'AICAR et la gemcitabine.
Les cibles du géfitinib et de l'AICAR sont tous deux les récepteurs du facteur de croissance épidermique (EGFR), et la gemcitabine est souvent utilisée pour traiter le cancer du poumon non à petites cellules sans mutations de l'EGFR.
Le document indique que, conformément aux modes d'action de ces médicaments, CODE-AE a constaté que les patients utilisant le géfitinib et l'AICAR présentaient des profils de réponse aux médicaments similaires.
En d’autres termes, CODE-AE a découvert la bonne cible pour le traitement des patients, c’est-à-dire qu’il peut prédire les médicaments applicables.
L'équipe de recherche ci-dessus appartient à la City University de New York.
L'auteur correspondant est Lei Xie, diplômé de l'Université des sciences et technologies de Chine avec une licence en physique des polymères.
Diplômé d'une maîtrise en informatique de l'Université Rutgers ; d'un doctorat de l'Université Rutgers, mais avec un diplôme en chimie.

Il est entendu que la prochaine étape de l'équipe de recherche consistera à développer la fonction de prédiction de CODE-AE en termes de concentration et de métabolisme de la réponse clinique de nouveaux médicaments.
Les chercheurs ont déclaré que le modèle d'IA pourrait également être adapté pour prédire les effets secondaires des médicaments sur le corps humain.
Il convient de mentionner que la sous-revue Nature "Nature Machine Intelligence" se concentre spécifiquement sur la recherche appliquée interdisciplinaire en intelligence artificielle et en sciences de la vie, avec un nombre moyen d'articles publiés chaque année d'environ 60.
Adresse papier : https://www.nature.com/articles/s42256-022-00541-0
Lien de référence : https://phys.org/news/2022-10-ai-accurately-human-response -drogue.html
Ce qui précède est le contenu détaillé de. pour plus d'informations, suivez d'autres articles connexes sur le site Web de PHP en chinois!
Articles Liés
Voir plus- Tendances technologiques à surveiller en 2023
- Comment l'intelligence artificielle apporte un nouveau travail quotidien aux équipes des centres de données
- L'intelligence artificielle ou l'automatisation peuvent-elles résoudre le problème de la faible efficacité énergétique des bâtiments ?
- Co-fondateur d'OpenAI interviewé par Huang Renxun : les capacités de raisonnement de GPT-4 n'ont pas encore atteint les attentes
- Bing de Microsoft surpasse Google en termes de trafic de recherche grâce à la technologie OpenAI