
 Technology peripheralsAIThe prediction accuracy is as high as 0.98. Tsinghua University, Shenzhen Technology and others proposed a multifunctional prediction framework for MOF materials based on Transformer.
Technology peripheralsAIThe prediction accuracy is as high as 0.98. Tsinghua University, Shenzhen Technology and others proposed a multifunctional prediction framework for MOF materials based on Transformer.

Editor| Out of huge potential.
Traditional simulation methods, such as molecular dynamics, although complex and computationally demanding, are highly accurate in simulating system behavior. In contrast, machine learning methods based on feature engineering perform better when dealing with complex systems. However, due to the scarcity of labeled data, it can easily lead to overfitting problems. Furthermore, these machine learning methods are usually designed to solve a single task and lack support for multi-task learning. Therefore, when choosing an appropriate method, factors such as accuracy, data requirements, and task complexity need to be weighed to find the solution that best fits the specific problem.
To address these challenges, a multi-institutional team composed of Tsinghua University, University of California, Sun Yat-sen University, Suzhou University, Shenzhen Technology and AI for Science Institute (AISI, Beijing) collaborated present Uni-MOF, an innovative framework for large-scale three-dimensional MOF representation learning, designed for multi-purpose gas prediction. Uni-MOFs are suitable for both scientific research and practical applications.
Uni-MOF can be regarded as a multifunctional gas adsorption predictor for MOF materials, showing excellent prediction accuracy in simulated data, marking an important application of machine learning in gas adsorption research.
The study was titled "A comprehensive transformer-based approach for high-accuracy gas adsorption predictions in metal-organic frameworks" and was published in "Nature Communications" on March 1, 2024.
 Paper link: https://www.nature.com/articles/s41467-024-46276-x
Paper link: https://www.nature.com/articles/s41467-024-46276-x
Need a unified adsorption framework
Metal-organic frameworks (MOFs) are widely used in gas separation and other fields because of their adjustable structural properties and chemical composition.
Although MOFs have great potential for gas adsorption, accurately predicting their adsorption capacity remains a challenge.
Computational methods such as molecular dynamics and Monte Carlo (MC) have high computational costs and complex implementation, which limits their use in large-scale, multi-gas and high-throughput calculations. In addition, gas adsorption operates over a wide range of conditions, making predictions more complex.
Graph neural networks and Transformers have been shown to successfully predict MOF properties.
Although existing models for predicting adsorption properties have high performance and strong predictive capabilities, they are usually designed for a single task, specifically predicting the adsorption absorption rate of a specific gas under specific conditions. However, the available datasets for these single-task predictions are often limited, hindering the generalizability of the models.
On the other hand, the combination of labeled data from various adsorbed gases in different temperature and pressure environments can create large data sets suitable for training across the entire operating conditions. The increased amount of data can also enhance the model's generalization capabilities and improve its practical industrial use. Therefore, a unified adsorption framework is needed to advance these models.
In addition, ensemble representation learning, or pre-training, for large-scale unlabeled MOF structures can further improve model performance and representation capabilities.
Uni-MOF Framework: Suitable for both scientific research and practical applications
Inspired by this, the research team proposed the Uni-MOF framework as a multi-purpose solution that uses structural representation learning to predict Gas adsorption of MOF under different conditions.
Compared with other Transformer-based models (such as MOFormer and MOFTransformer), Uni-MOF, as a Transformer-based framework, can not only identify and restore the three-dimensional structure of nanoporous materials in pre-training, thus greatly improving Robustness of nanoporous materials. And the fine-tuning task further takes into account operating conditions such as temperature, pressure and different gas molecules, making Uni-MOF suitable for both scientific research and practical applications.
Uni-MOF As a comprehensive gas adsorption estimator for MOF materials, only the crystal information file (CIF) of the MOF and related gas, temperature and pressure parameters are needed to predict nanoporous materials under a wide range of operating conditions. gas adsorption characteristics. The Uni-MOF framework is easy to use and allows module selection.
In addition, the problem of overfitting is effectively solved by combining various cross-system absorption labeled data with representation learning of a large amount of unlabeled structural data. This compensates for both high-quality data and data deficiencies, ultimately improving the accuracy of gas adsorption predictions.
The Uni-MOF framework enables atomic-level material identification accuracy, while integrated models make Uni-MOF more applicable to engineering problems. There is no doubt that achieving truly unified models is the future direction of the materials field, rather than just focusing on specialized fields. Uni-MOF is a pioneering practice of machine learning in the field of gas adsorption.
Uni-MOF Framework Overview
The Uni-MOF framework includes pre-training of three-dimensional nanoporous crystals and fine-tuning of multi-task predictions for downstream applications.

Figure 1: Schematic of the Uni-MOF framework. (Source: Paper)
Pre-training on 3D crystalline materials significantly enhances the prediction performance for downstream tasks, especially for large-scale unlabeled data.
To solve the problem of insufficient supervision of training data sets, researchers collected a large dataset of MOF structures and generated more than 300,000 MOFs using ToBaCCo.3.0. High-throughput construction of COFs based on Materials Genome Strategies and Quasi-Reactive Assembly Algorithm (QReaxAA) is feasible to establish a comprehensive COF library. Through the spatial configuration of the material, Uni-MOF is able to well learn the structural properties of the material, and the most important thing is the chemical bond information.
In order to enable Uni-MOF to learn a more diverse range of materials and thus improve the generalization ability to a wider range of materials, MOF and COF were introduced virtually and experimentally during the pre-training process. Similar to the masked labeling task in BERT and Uni-Mol, Uni-MOF adopts the prediction task of masked atoms, thereby facilitating pre-trained models to gain in-depth understanding of the material spatial structure.
To enhance the robustness of pre-training and generalize the learned representations, the researchers introduced noise to the original coordinates of MOFs. In the pre-training phase, two tasks are designed. (1) Reconstruct original 3D positions from noisy data, and (2) predict shielded atoms. These tasks can enhance model robustness and improve downstream predictive performance.
In addition to diverse spatial configurations, a comprehensive set of material property data points is also critical for model training. To enrich the dataset, the researchers established a custom data generation process (shown in Figure 1b).
Fine-tuning of Uni-MOF is based on the extraction of representations obtained through pre-training, and the use of home-made workflows to generate and collect large datasets. During the fine-tuning process, approximately 3,000,000 labeled data points under various adsorption conditions for MOFs and COFs were used to train the model, enabling accurate prediction of adsorption capacity.
With a diverse database of cross-system target data, Uni-MOF is fine-tuned to predict the multi-system adsorption properties of MOFs in any state. Therefore, Uni-MOF is a unified and easy-to-use framework for predicting the adsorption performance of MOF adsorbents.
Most importantly, Uni-MOF requires no additional labor to identify human-defined structural features. Instead, the CIF of the MOF and the associated gas, temperature and pressure parameters are sufficient. The self-supervised learning strategy and rich database ensure that Uni-MOF is able to predict the gas adsorption properties of nanoporous materials under various operating parameters, making it a proficient estimator of gas adsorption for MOF materials.
Prediction accuracy up to 0.98, predicts across systems
The study performed self-supervised learning on a database of more than 631,000 MOFs and COFs, with prediction accuracy up to 0.98. This shows that the representation learning framework based on 3D pre-training effectively learns the complex structural information of MOF while avoiding overfitting.
Uni-MOF was applied to predict the gas adsorption performance of three major databases (hMOF_MOFX-DB, CoRE_MOFX-DB and CoRE_MAP_DB), and a prediction accuracy of up to 0.98 was achieved in databases with sufficient data.

#Figure 2: Overall performance of Uni-MOF in large-scale databases. (Source: paper)
When the data set is fully sampled, Uni-MOF not only maintains a prediction accuracy of more than 0.83, but also can accurately select high performance at high pressure by predicting adsorption at low pressure only. adsorbent, consistent with the experimental screening results. Uni-MOF therefore represents a major breakthrough in the application of machine learning techniques in the field of materials science.

Figure 3: Adsorption isotherms based on low pressure predictions and high pressure experimental values, each curve represents a Langmuir fit. (Source: paper)
In addition, compared with single-system tasks, the Uni-MOF framework shows superior performance on cross-system data sets and can accurately predict the adsorption characteristics of unknown gases with a prediction accuracy as high as 0.85, Demonstrates its strong predictive power and versatility.

Figure 4: Uni-MOF cross-system prediction case. (Source: paper)
Research shows that pre-trained self-supervised learning strategies can effectively improve the robustness and downstream prediction performance of Uni-MOF.

Figure 5: Comparison of Uni-MOF and Uni-MOF without pre-training. (Source: paper)
Through extensive pre-training on three-dimensional structures, Uni-MOF effectively learns the structural features of MOFs and achieves a high coefficient of determination of 0.99 for hMOFs.

Figure 6: Prediction and analysis of structural characteristics. (Source: paper)
In addition, t-SNE (t-distributed stochastic neighbor embedding) analysis confirmed that the fine-tuning stage can further learn structural features and can well identify structures with different adsorbate behaviors, which It is shown that there is a strong correlation between the learned representation and the gas adsorption target.

Figure 7: Visualization of MOF structural representation in hMOF and CoRE_MOF datasets, low-dimensional embeddings computed by t-SNE method. (Source: paper)
In short, the Uni-MOF framework, as a multi-functional prediction platform for MOF materials, acts as a gas adsorption estimator for MOFs and has high accuracy in predicting gas adsorption under different operating conditions. It has broad application prospects in the field of materials science.
The above is the detailed content of The prediction accuracy is as high as 0.98. Tsinghua University, Shenzhen Technology and others proposed a multifunctional prediction framework for MOF materials based on Transformer.. For more information, please follow other related articles on the PHP Chinese website!

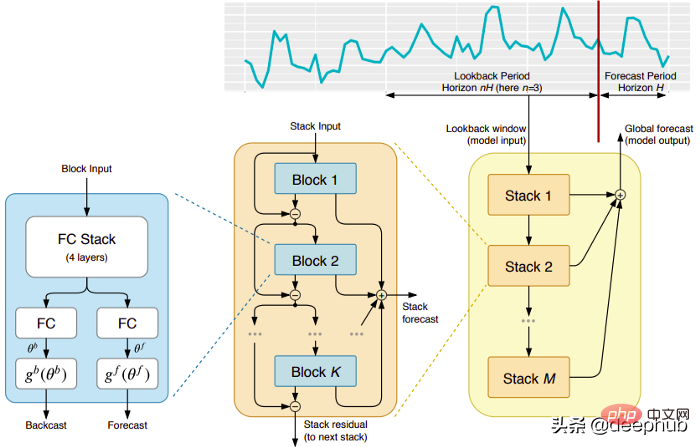 五个时间序列预测的深度学习模型对比总结May 05, 2023 pm 05:16 PM
五个时间序列预测的深度学习模型对比总结May 05, 2023 pm 05:16 PMMakridakisM-Competitions系列(分别称为M4和M5)分别在2018年和2020年举办(M6也在今年举办了)。对于那些不了解的人来说,m系列得比赛可以被认为是时间序列生态系统的一种现有状态的总结,为当前得预测的理论和实践提供了经验和客观的证据。2018年M4的结果表明,纯粹的“ML”方法在很大程度上胜过传统的统计方法,这在当时是出乎意料的。在两年后的M5[1]中,最的高分是仅具有“ML”方法。并且所有前50名基本上都是基于ML的(大部分是树型模型)。这场比赛看到了LightG
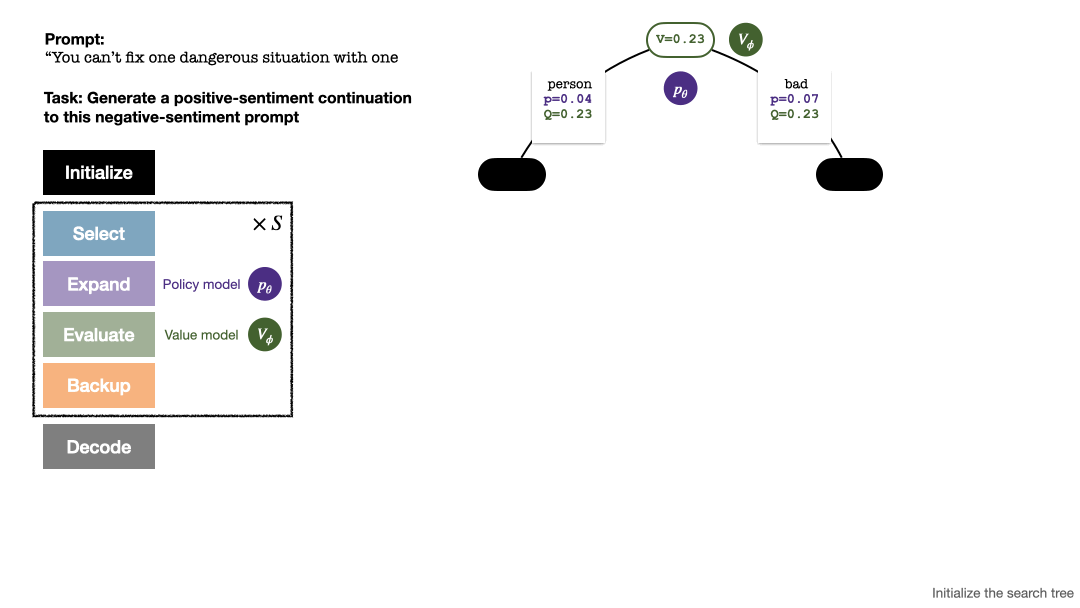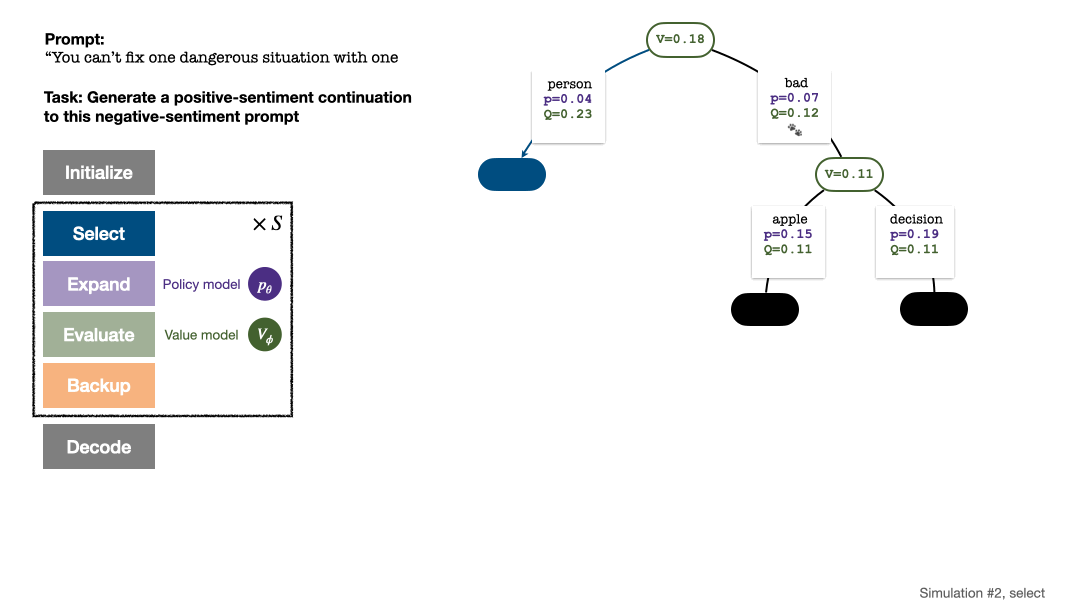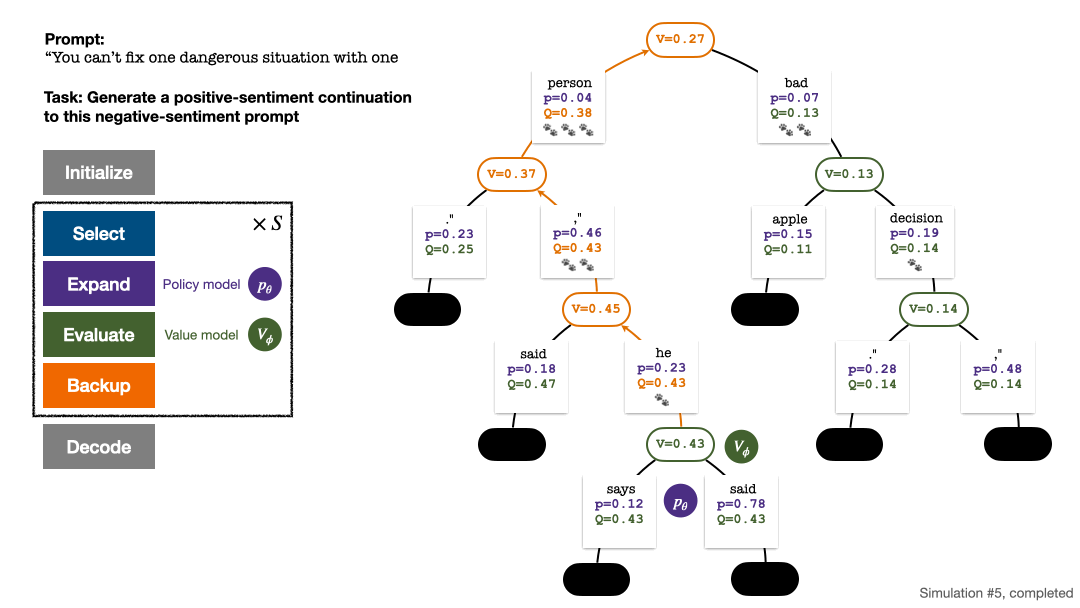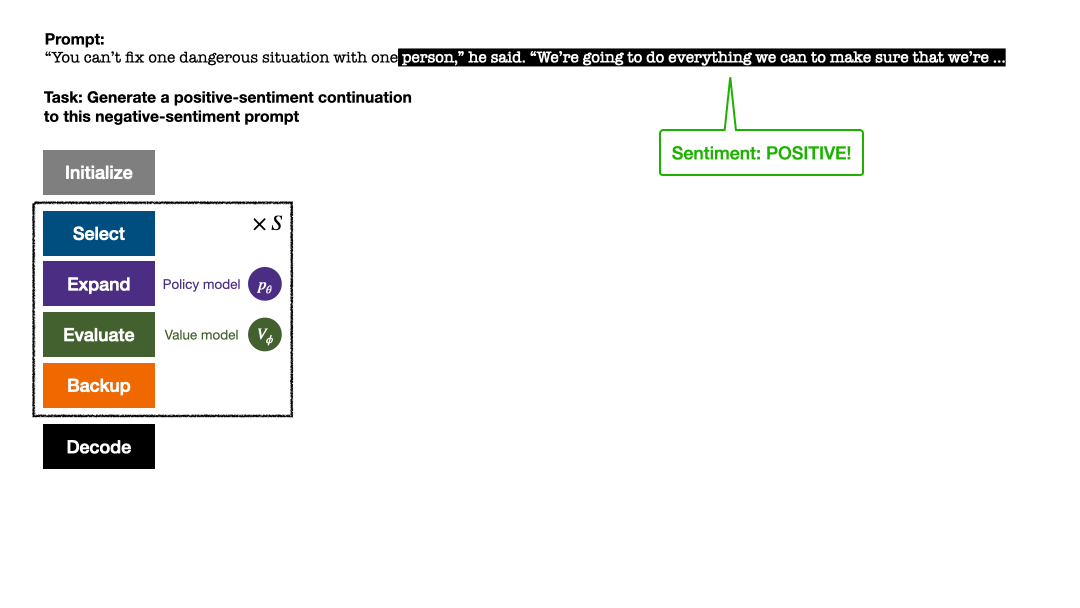 RLHF与AlphaGo核心技术强强联合,UW/Meta让文本生成能力再上新台阶Oct 27, 2023 pm 03:13 PM
RLHF与AlphaGo核心技术强强联合,UW/Meta让文本生成能力再上新台阶Oct 27, 2023 pm 03:13 PM在一项最新的研究中,来自UW和Meta的研究者提出了一种新的解码算法,将AlphaGo采用的蒙特卡洛树搜索算法(Monte-CarloTreeSearch,MCTS)应用到经过近端策略优化(ProximalPolicyOptimization,PPO)训练的RLHF语言模型上,大幅提高了模型生成文本的质量。PPO-MCTS算法通过探索与评估若干条候选序列,搜索到更优的解码策略。通过PPO-MCTS生成的文本能更好满足任务要求。论文链接:https://arxiv.org/pdf/2309.150
 MIT团队运用机器学习闭环自主分子发现平台,成功发现、合成和描述了303种新分子Jan 04, 2024 pm 05:38 PM
MIT团队运用机器学习闭环自主分子发现平台,成功发现、合成和描述了303种新分子Jan 04, 2024 pm 05:38 PM编辑|X传统意义上,发现所需特性的分子过程一直是由手动实验、化学家的直觉以及对机制和第一原理的理解推动的。随着化学家越来越多地使用自动化设备和预测合成算法,自主研究设备越来越接近实现。近日,来自MIT的研究人员开发了由集成机器学习工具驱动的闭环自主分子发现平台,以加速具有所需特性的分子的设计。无需手动实验即可探索化学空间并利用已知的化学结构。在两个案例研究中,该平台尝试了3000多个反应,其中1000多个产生了预测的反应产物,提出、合成并表征了303种未报道的染料样分子。该研究以《Autonom
 AI助力脑机接口研究,纽约大学突破性神经语音解码技术,登Nature子刊Apr 17, 2024 am 08:40 AM
AI助力脑机接口研究,纽约大学突破性神经语音解码技术,登Nature子刊Apr 17, 2024 am 08:40 AM作者|陈旭鹏编辑|ScienceAI由于神经系统的缺陷导致的失语会导致严重的生活障碍,它可能会限制人们的职业和社交生活。近年来,深度学习和脑机接口(BCI)技术的飞速发展为开发能够帮助失语者沟通的神经语音假肢提供了可行性。然而,神经信号的语音解码面临挑战。近日,约旦大学VideoLab和FlinkerLab的研究者开发了一个新型的可微分语音合成器,可以利用一个轻型的卷积神经网络将语音编码为一系列可解释的语音参数(例如音高、响度、共振峰频率等),并通过可微分神经网络将这些参数合成为语音。这个合成器
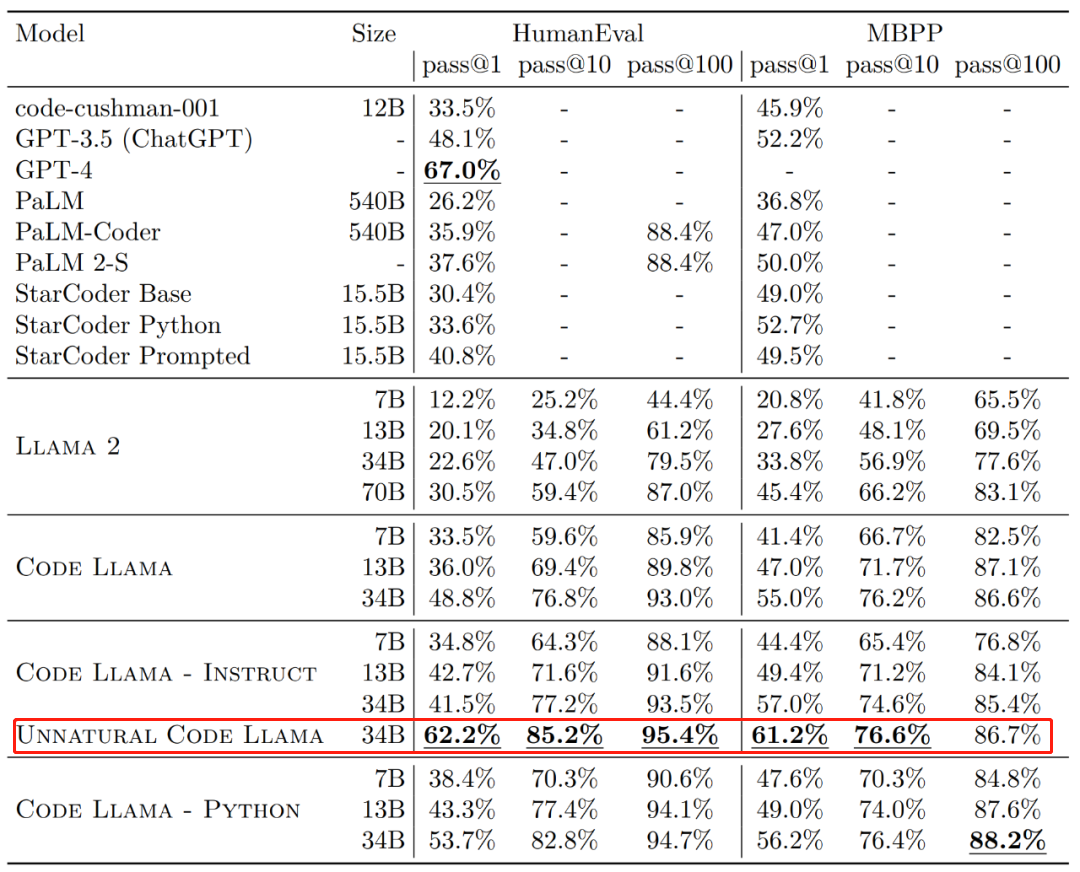 Code Llama代码能力飙升,微调版HumanEval得分超越GPT-4,一天发布Aug 26, 2023 pm 09:01 PM
Code Llama代码能力飙升,微调版HumanEval得分超越GPT-4,一天发布Aug 26, 2023 pm 09:01 PM昨天,Meta开源专攻代码生成的基础模型CodeLlama,可免费用于研究以及商用目的。CodeLlama系列模型有三个参数版本,参数量分别为7B、13B和34B。并且支持多种编程语言,包括Python、C++、Java、PHP、Typescript(Javascript)、C#和Bash。Meta提供的CodeLlama版本包括:代码Llama,基础代码模型;代码羊-Python,Python微调版本;代码Llama-Instruct,自然语言指令微调版就其效果来说,CodeLlama的不同版
 手机摄影技术让以假乱真的好莱坞级电影特效视频走红Sep 07, 2023 am 09:41 AM
手机摄影技术让以假乱真的好莱坞级电影特效视频走红Sep 07, 2023 am 09:41 AM一个普通人用一台手机就能制作电影特效的时代已经来了。最近,一个名叫Simulon的3D技术公司发布了一系列特效视频,视频中的3D机器人与环境无缝融合,而且光影效果非常自然。呈现这些效果的APP也叫Simulon,它能让使用者通过手机摄像头的实时拍摄,直接渲染出CGI(计算机生成图像)特效,就跟打开美颜相机拍摄一样。在具体操作中,你要先上传一个3D模型(比如图中的机器人)。Simulon会将这个模型放置到你拍摄的现实世界中,并使用准确的照明、阴影和反射效果来渲染它们。整个过程不需要相机解算、HDR
 准确率 >98%,基于电子密度的 GPT 用于化学研究,登 Nature 子刊Mar 27, 2024 pm 02:16 PM
准确率 >98%,基于电子密度的 GPT 用于化学研究,登 Nature 子刊Mar 27, 2024 pm 02:16 PM编辑|紫罗可合成分子的化学空间是非常广阔的。有效地探索这个领域需要依赖计算筛选技术,比如深度学习,以便快速地发现各种有趣的化合物。将分子结构转换为数字表示形式,并开发相应算法生成新的分子结构是进行化学发现的关键。最近,英国格拉斯哥大学的研究团队提出了一种基于电子密度训练的机器学习模型,用于生成主客体binders。这种模型能够以简化分子线性输入规范(SMILES)格式读取数据,准确率高达98%,从而实现对分子在二维空间的全面描述。通过变分自编码器生成主客体系统的电子密度和静电势的三维表示,然后通
 NVIDIA、Mila、Caltech联合发布LLM结合药物发现的多模态分子结构-文本模型Jan 14, 2024 pm 08:00 PM
NVIDIA、Mila、Caltech联合发布LLM结合药物发现的多模态分子结构-文本模型Jan 14, 2024 pm 08:00 PM作者|刘圣超编辑|凯霞从2021年开始,大语言和多模态的结合席卷了机器学习科研界。随着大模型和多模态应用的发展,我们是否可以将这些技术应用于药物发现呢?而且,这些自然语言的文本描述是否可以为这个具有挑战性的问题带来新的视角呢?答案是肯定的,并且我们对此持乐观态度近日,加拿大蒙特利尔学习算法研究院(Mila)、NVIDIAResearch、伊利诺伊大学厄巴纳-香槟分校(UIUC)、普林斯顿大学和加州理工学院的研究团队,通过对比学习策略共同学习分子的化学结构和文本描述,提出了一种多模态分子结构-文本


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

EditPlus Chinese cracked version
Small size, syntax highlighting, does not support code prompt function

SublimeText3 Linux new version
SublimeText3 Linux latest version

ZendStudio 13.5.1 Mac
Powerful PHP integrated development environment

Notepad++7.3.1
Easy-to-use and free code editor

SublimeText3 English version
Recommended: Win version, supports code prompts!

