
 Technology peripheralsAIPredicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery
Technology peripheralsAIPredicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery

Post-stage functionalization is an economical method for optimizing the properties of drug candidates. However, the chemical complexity of drug molecules often makes late-stage functionalization challenging
To solve this problem, researchers from the University of Munich, ETH Zurich, and Roche Basel collaborated to develop a late-stage Functionalization platform, which is based on geometric deep learning and high-throughput reaction screening technology
Given that borylation is one of the key steps of functionalization, we used computational models to predict the yield under different reaction conditions, The average absolute error range is 4-5%. The model was able to classify new reactions for known and unknown substrates with an accuracy of 92% and 67% respectively. We were able to accurately capture the regioselectivity of the main product, with an F-score of 67% for the classifier. We successfully identified many opportunities for structural diversification when applied to 23 different commercial drug molecules
The study is titled "Using Geometric Deep Learning to Enable High-Throughput Experiments to Promote Late-stage Drug Diversification", Published in the journal Nature Chemistry on November 23, 2023

LSF projects play an important role in medicinal chemistry research
The novelty and complexity of structures make synthesizing chemical target structures challenging when aiming to establish structure-activity relationships in medicinal chemistry. Structure-activity relationship models can guide lead compounds and lead compound optimization plans to improve the pharmacological activity and physicochemical properties of drug candidates. Efficient integration is crucial for the exploration of structure-activity relationships, which is the bottleneck of the design-make-test-analyze cycle
There are many alternative methods to activate and modify C-H bonds to achieve organic scaffolds Late Stage Functionalization (LSF), ranging from molecular building blocks to advanced drug molecules. Many catalytic systems provide directional and non-directional approaches, as well as chemical and site-selective access to modified analogues
Among the numerous LSF methods, the C-H borylation method is considered the most commonly used for fast compounds Diversify your approach. Organoboron compounds can be converted into a variety of functional groups as a reliable means for subsequent C-C bond coupling reactions, thereby enabling extensive structure-activity relationship research
However, currently, in drug discovery, LSF’s The app has only a few reports. Most of these reports focus on a single LSF reaction type. Direct LSF of multiple types of C–H bonds with different bond strengths, electronic properties, and steric and functional group environments poses challenges. Furthermore, conducting LSF projects is often time and resource intensive, which is not consistent with the tight schedules and limited assets of many medicinal chemistry projects

Diagram showing boronation An overview of diversity research. (Data source: paper)
Artificial Intelligence Supported LSF (Language Support Feature)
High-throughput experiment (HTE) is an established reaction optimization method. Semi-automated, miniaturized low-batch screening is possible, allowing multiple transformations to be performed in parallel using a small number of precious building blocks and consumables quickly and reproducibly. Combined with FAIR (Findability, Accessibility, Interoperability, Reusability) documentation that generates high-quality datasets on successful and failed responses, HTE enables advanced data analytics and machine learning to unlock LSF for drug discovery. Foundation.
Graph neural network (GNN) has been widely used in molecular feature extraction and attribute prediction. Among various machine learning methods developed for chemical reaction planning, GNNs have been successfully applied to retrosynthetic planning, regioselectivity prediction, and reaction product prediction. In addition, transformers and fingerprint-based methods have also been developed to solve similar problems
Research has shown that by learning the geometry of the transition state, the outcome of a competing reaction can be accurately predicted. Predictions of the regioselectivity of reactions driven by electronic effects can be improved using density functional theory (DFT) and graphical characterization of atomic partial charges. Combining graph machine learning and high-throughput experiments (HTE), conditions for C-H activation reactions of organic substrates can be optimized. Some studies have focused on the use of deep learning models of transition states that have the ability to predict reaction outcomes, including enantioselectivity in some cases. However, these methods are limited to small molecule structures and relatively small molecules. The small data set makes it challenging to apply such models to more structurally complex drug-like molecules. Based on literature research, the regioselectivity of iridium-catalyzed borylation reactions can be predicted through a hybrid machine learning model enhanced with quantum chemical information of transition states. However, the impact of steric and electronic effects has not yet been explored on the performance of C-H activation reaction models and their regioselective applications in molecules with multiple aromatic ring systems
Automated LSF Boron Screening with Geometric Deep Learning
Researchers from the University of Munich, ETH Zurich and Roche Pharmaceuticals Basel present an automatic LSF boron screening method applied to geometric deep learning LSF boron screening method for identifying late-stage hits and lead diversification opportunities. Computational deep learning was employed to predict reaction outcome, yield, and regioselectivity of complex drug molecules LSF.

With this approach, the number of laboratory experiments could be significantly reduced, leading to a higher Efficiency and Sustainability of Chemical Synthesis
For the first step of this research, we conducted a thorough analysis of the published literature in order to select appropriate reaction conditions for high-throughput screening and late-stage lead compounds for drug discovery Substrates with relevant properties. We determined the reaction conditions based on a manually organized data set of 38 literatures
The selection of LSF substrates was based on the results of cluster analysis of 1,174 approved drugs, resulting in 23 structurally different drug molecules. This approach enables researchers to use relevant examples of reaction conditions and substrates in an "information library" approach, rather than relying solely on ideal substrates and fragments of limited applicability to optimize the synthesis of lead compounds
In the second step, researchers generate data (experimental datasets) using semi-automated high-throughput experiments (HTE). The reaction data of selected drug molecules and reaction conditions provide high-quality data for machine learning of subsequent reaction results
Finally, we trained different graph neural network (GNN) models using two-dimensional, three-dimensional and Molecular maps with enhanced partial charges on atoms to predict binary reaction outcomes, reaction yields, and regioselectivities. "Interestingly, the predictions improved when we considered three-dimensional information about the starting material rather than just its two-dimensional chemical formula," said Kenneth Atz, a PhD student at ETH Zurich. The method has been successfully used to identify positions in existing active ingredients where additional reactive groups can be introduced. This will help researchers more quickly develop new, more effective variants of known pharmaceutical active ingredients
Please click the following link to view the paper content: https://www.nature.com/articles/ s41557-023-01360-5
Related reports: https://techxplore.com/news/2023-11-Artificial intelligence paves the way for drugs.html
The above is the detailed content of Predicting optimal options for synthesizing drug molecules using geometric deep learning methods, paving the way for new drug discovery. For more information, please follow other related articles on the PHP Chinese website!

 What is Graph of Thought in Prompt EngineeringApr 13, 2025 am 11:53 AM
What is Graph of Thought in Prompt EngineeringApr 13, 2025 am 11:53 AMIntroduction In prompt engineering, “Graph of Thought” refers to a novel approach that uses graph theory to structure and guide AI’s reasoning process. Unlike traditional methods, which often involve linear s
 Optimize Your Organisation's Email Marketing with GenAI AgentsApr 13, 2025 am 11:44 AM
Optimize Your Organisation's Email Marketing with GenAI AgentsApr 13, 2025 am 11:44 AMIntroduction Congratulations! You run a successful business. Through your web pages, social media campaigns, webinars, conferences, free resources, and other sources, you collect 5000 email IDs daily. The next obvious step is
 Real-Time App Performance Monitoring with Apache PinotApr 13, 2025 am 11:40 AM
Real-Time App Performance Monitoring with Apache PinotApr 13, 2025 am 11:40 AMIntroduction In today’s fast-paced software development environment, ensuring optimal application performance is crucial. Monitoring real-time metrics such as response times, error rates, and resource utilization can help main
 ChatGPT Hits 1 Billion Users? 'Doubled In Just Weeks' Says OpenAI CEOApr 13, 2025 am 11:23 AM
ChatGPT Hits 1 Billion Users? 'Doubled In Just Weeks' Says OpenAI CEOApr 13, 2025 am 11:23 AM“How many users do you have?” he prodded. “I think the last time we said was 500 million weekly actives, and it is growing very rapidly,” replied Altman. “You told me that it like doubled in just a few weeks,” Anderson continued. “I said that priv
 Pixtral-12B: Mistral AI's First Multimodal Model - Analytics VidhyaApr 13, 2025 am 11:20 AM
Pixtral-12B: Mistral AI's First Multimodal Model - Analytics VidhyaApr 13, 2025 am 11:20 AMIntroduction Mistral has released its very first multimodal model, namely the Pixtral-12B-2409. This model is built upon Mistral’s 12 Billion parameter, Nemo 12B. What sets this model apart? It can now take both images and tex
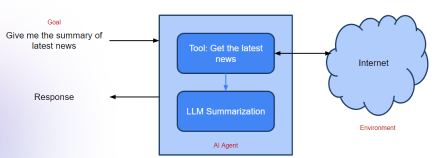 Agentic Frameworks for Generative AI Applications - Analytics VidhyaApr 13, 2025 am 11:13 AM
Agentic Frameworks for Generative AI Applications - Analytics VidhyaApr 13, 2025 am 11:13 AMImagine having an AI-powered assistant that not only responds to your queries but also autonomously gathers information, executes tasks, and even handles multiple types of data—text, images, and code. Sounds futuristic? In this a
 Applications of Generative AI in the Financial SectorApr 13, 2025 am 11:12 AM
Applications of Generative AI in the Financial SectorApr 13, 2025 am 11:12 AMIntroduction The finance industry is the cornerstone of any country’s development, as it drives economic growth by facilitating efficient transactions and credit availability. The ease with which transactions occur and credit
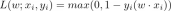 Guide to Online Learning and Passive-Aggressive AlgorithmsApr 13, 2025 am 11:09 AM
Guide to Online Learning and Passive-Aggressive AlgorithmsApr 13, 2025 am 11:09 AMIntroduction Data is being generated at an unprecedented rate from sources such as social media, financial transactions, and e-commerce platforms. Handling this continuous stream of information is a challenge, but it offers an


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

Atom editor mac version download
The most popular open source editor

ZendStudio 13.5.1 Mac
Powerful PHP integrated development environment

SublimeText3 Chinese version
Chinese version, very easy to use

WebStorm Mac version
Useful JavaScript development tools

VSCode Windows 64-bit Download
A free and powerful IDE editor launched by Microsoft

