

 Technology peripheralsAITsinghua team proposes knowledge-guided graph Transformer pre-training framework: a method to improve molecular representation learning
Technology peripheralsAITsinghua team proposes knowledge-guided graph Transformer pre-training framework: a method to improve molecular representation learningTsinghua team proposes knowledge-guided graph Transformer pre-training framework: a method to improve molecular representation learning


In order to facilitate molecular property prediction, in the field of drug discovery, it is very important to learn effective molecular feature representations. Recently, people have overcome the challenge of data scarcity by pre-training graph neural networks (GNN) using self-supervised learning techniques. However, there are two main problems with current methods based on self-supervised learning: the lack of clear self-supervised learning strategies and the limited capabilities of GNN
Recently, a research team from Tsinghua University, West Lake University and Zhijiang Laboratory, We propose Knowledge-guided Pre-training of Graph Transformer (KPGT), a self-supervised learning framework that provides improved, generalizable and robust learning through significantly enhanced molecular representation learning. Molecular property prediction. The KPGT framework integrates a graph Transformer designed specifically for molecular graphs and a knowledge-guided pre-training strategy to fully capture the structural and semantic knowledge of molecules.
Through extensive computational testing on 63 data sets, KPGT has demonstrated superior performance in predicting molecular properties in various fields. Furthermore, the practical applicability of KPGT in drug discovery was verified by identifying potential inhibitors of two antitumor targets. Overall, KPGT can provide a powerful and useful tool for advancing the AI-assisted drug discovery process.
The research was titled "A knowledge-guided pre-training framework for improving molecular representation learning" and was published in "Nature Communications" on November 21, 2023.

Determining molecular properties experimentally requires significant time and resources, and identifying molecules with desired properties is one of the most significant challenges in drug discovery. In recent years, artificial intelligence-based methods have played an increasingly important role in predicting molecular properties. One of the main challenges of artificial intelligence-based methods for predicting molecular properties is the characterization of molecules
In recent years, the emergence of deep learning-based methods has emerged as a potentially useful tool for predicting molecular properties, mainly because they have the ability to transform from simple inputs Excellent ability to automatically extract effective features from data. Notably, various neural network architectures, including recurrent neural networks (RNN), convolutional neural networks (CNN), and graph neural networks (GNN), are adept at modeling molecular data in various formats, ranging from simplified molecular inputs to Line input system (SMILES) to molecular images and molecular diagrams. However, the limited availability of marker molecules and the vastness of chemical space limit their predictive performance, especially when dealing with out-of-distribution data samples.
With the remarkable achievements of self-supervised learning methods in the fields of natural language processing and computer vision, these techniques have been applied to pre-train GNNs and improve representation learning of molecules, thereby achieving success in downstream molecular property prediction tasks. Substantial progress has been made
The researchers hypothesize that introducing additional knowledge that quantitatively describes molecular characteristics into a self-supervised learning framework can effectively address these challenges. Molecules have many quantitative characteristics, such as molecular descriptors and fingerprints, that can be easily obtained with currently established computational tools. Integrating this additional knowledge can introduce rich molecular semantic information into self-supervised learning, thereby greatly enhancing the acquisition of semantically rich molecular representations.
Generally, existing self-supervised learning methods rely on GNN as the core model. However, GNN has limited model capacity. Furthermore, GNNs can have difficulty capturing long-range interactions between atoms. And Transformer-based models have become a game-changing model. It is characterized by an increasing number of parameters and the ability to capture long-range interactions, providing a promising approach to comprehensively simulating the structural characteristics of molecules
Self-supervised learning framework KPGT
In this study, the researchers introduced a self-supervised learning framework called KPGT, which aims to enhance molecular representation learning to promote downstream molecular property prediction tasks. The KPGT framework consists of two main components: a backbone model called Line Graph Transformer (LiGhT) and a knowledge-guided pre-training policy. The KPGT framework combines the high-capacity LiGhT model, which is specifically designed to accurately model molecular graph structures, and utilizes a knowledge-guided pre-training strategy to capture molecular structure and semantic knowledge
The research team used About 2 million molecules, LiGhT was pre-trained through a knowledge-guided pre-training strategy
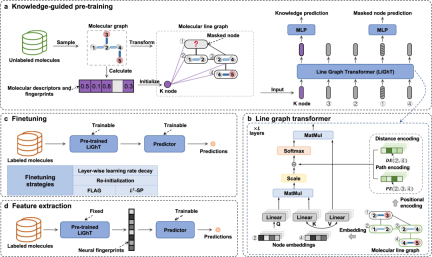
Rewritten content: Figure: KPGT Overview. (Source: paper)
KPGT outperforms baseline methods in molecular property prediction. Compared with several baseline methods, KPGT achieves significant improvements on 63 datasets.
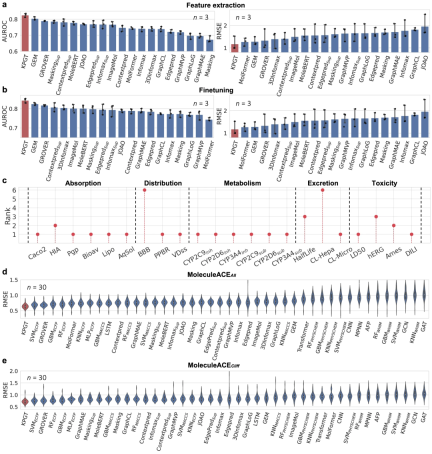
In addition, by successfully using KPGT to identify potential inhibitors of two anti-tumor targets, hematopoietic progenitor kinase 1 (HPK1) and fibroblast growth factor receptor (FGFR1), it was demonstrated Practical applicability of KPGT.

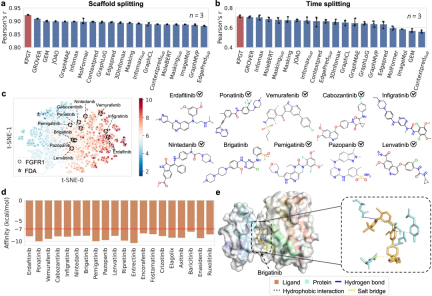
Research Limitations
Despite the advantages of KPGT in effective molecular property prediction, there are still some limitations.
First of all, the integration of additional knowledge is the most significant feature of the proposed method. In addition to the 200 molecular descriptors and 512 RDKFPs used in KPGT, there is the potential to incorporate various other types of additional information knowledge. Additionally, further research could integrate three-dimensional (3D) molecular conformation into the pre-training process, allowing the model to capture important 3D information about the molecule and potentially enhance representation learning. ability. While KPGT currently employs a backbone model with approximately 100 million parameters and pre-training on 2 million molecules, exploring larger-scale pre-training can provide insights into molecular representation learning. More substantial benefits.
Overall, KPGT provides a powerful self-supervised learning framework for effective molecular representation learning, thereby advancing the field of artificial intelligence-assisted drug discovery.
Paper link: https://www.nature.com/articles/s41467-023-43214-1
The above is the detailed content of Tsinghua team proposes knowledge-guided graph Transformer pre-training framework: a method to improve molecular representation learning. For more information, please follow other related articles on the PHP Chinese website!

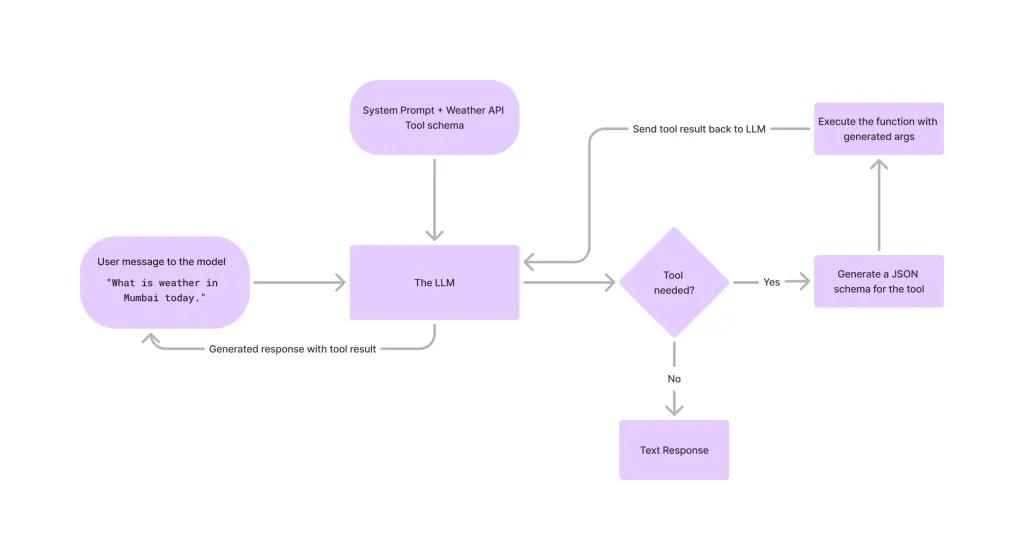 Tool Calling in LLMsApr 14, 2025 am 11:28 AM
Tool Calling in LLMsApr 14, 2025 am 11:28 AMLarge language models (LLMs) have surged in popularity, with the tool-calling feature dramatically expanding their capabilities beyond simple text generation. Now, LLMs can handle complex automation tasks such as dynamic UI creation and autonomous a
 How ADHD Games, Health Tools & AI Chatbots Are Transforming Global HealthApr 14, 2025 am 11:27 AM
How ADHD Games, Health Tools & AI Chatbots Are Transforming Global HealthApr 14, 2025 am 11:27 AMCan a video game ease anxiety, build focus, or support a child with ADHD? As healthcare challenges surge globally — especially among youth — innovators are turning to an unlikely tool: video games. Now one of the world’s largest entertainment indus
 UN Input On AI: Winners, Losers, And OpportunitiesApr 14, 2025 am 11:25 AM
UN Input On AI: Winners, Losers, And OpportunitiesApr 14, 2025 am 11:25 AM“History has shown that while technological progress drives economic growth, it does not on its own ensure equitable income distribution or promote inclusive human development,” writes Rebeca Grynspan, Secretary-General of UNCTAD, in the preamble.
 Learning Negotiation Skills Via Generative AIApr 14, 2025 am 11:23 AM
Learning Negotiation Skills Via Generative AIApr 14, 2025 am 11:23 AMEasy-peasy, use generative AI as your negotiation tutor and sparring partner. Let’s talk about it. This analysis of an innovative AI breakthrough is part of my ongoing Forbes column coverage on the latest in AI, including identifying and explaining
 TED Reveals From OpenAI, Google, Meta Heads To Court, Selfie With MyselfApr 14, 2025 am 11:22 AM
TED Reveals From OpenAI, Google, Meta Heads To Court, Selfie With MyselfApr 14, 2025 am 11:22 AMThe TED2025 Conference, held in Vancouver, wrapped its 36th edition yesterday, April 11. It featured 80 speakers from more than 60 countries, including Sam Altman, Eric Schmidt, and Palmer Luckey. TED’s theme, “humanity reimagined,” was tailor made
 Joseph Stiglitz Warns Of The Looming Inequality Amid AI Monopoly PowerApr 14, 2025 am 11:21 AM
Joseph Stiglitz Warns Of The Looming Inequality Amid AI Monopoly PowerApr 14, 2025 am 11:21 AMJoseph Stiglitz is renowned economist and recipient of the Nobel Prize in Economics in 2001. Stiglitz posits that AI can worsen existing inequalities and consolidated power in the hands of a few dominant corporations, ultimately undermining economic
 What is Graph Database?Apr 14, 2025 am 11:19 AM
What is Graph Database?Apr 14, 2025 am 11:19 AMGraph Databases: Revolutionizing Data Management Through Relationships As data expands and its characteristics evolve across various fields, graph databases are emerging as transformative solutions for managing interconnected data. Unlike traditional
 LLM Routing: Strategies, Techniques, and Python ImplementationApr 14, 2025 am 11:14 AM
LLM Routing: Strategies, Techniques, and Python ImplementationApr 14, 2025 am 11:14 AMLarge Language Model (LLM) Routing: Optimizing Performance Through Intelligent Task Distribution The rapidly evolving landscape of LLMs presents a diverse range of models, each with unique strengths and weaknesses. Some excel at creative content gen


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

PhpStorm Mac version
The latest (2018.2.1) professional PHP integrated development tool

SublimeText3 English version
Recommended: Win version, supports code prompts!

WebStorm Mac version
Useful JavaScript development tools

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

Zend Studio 13.0.1
Powerful PHP integrated development environment

