
 Technology peripheralsAICharge-based atomic simulation implementation using pre-trained general purpose neural network CHGNet
Technology peripheralsAICharge-based atomic simulation implementation using pre-trained general purpose neural network CHGNetCharge-based atomic simulation implementation using pre-trained general purpose neural network CHGNet


Rewritten content is: Ziluo
Large-scale simulations of complex electron interactions remain one of the greatest challenges in atomic modeling. While classical force fields often fail to describe couplings between electronic states and ion rearrangements, more accurate ab initio molecular dynamics suffer from computational complexity that prevents long and large-scale simulations, which are relevant for research techniques The phenomenon is crucial
Recently, researchers from the University of California, Berkeley, and Lawrence Berkeley National Laboratory proposed a machine learning interatomic potential (MLIP) model based on graph neural networks: the crystalline Hamiltonian graph Neural network (Crystal Hamiltonian Graph Neural Network, CHGNet) can model a universal potential energy surface.
The study highlights the importance of charge information for capturing appropriate chemical reactions and provides insights into ion systems with additional electronic degrees of freedom unobservable with previous MLIPs.
The research was titled "CHGNet as a pretrained universal neural network potential for charge-informed atomistic modeling" and was published in "Nature Machine Intelligence" on September 14, 2023.

Large-scale simulations, such as molecular dynamics (MD), are important tools for computational exploration of solid-state materials. However, accurately modeling electronic interactions and their subtle effects in molecular dynamics simulations remains a huge challenge. Empirical methods such as classical force fields are often not accurate enough to capture complex electronic interactions
Ab initio molecular dynamics (AIMD) combined with density functional theory (DFT) can be used to explicitly calculate density functional Approximating the electronic structure within, producing high-fidelity results with quantum mechanical precision. Long-term, large-scale spin-polarized AIMD simulations, critical for studying ion migration, phase transitions, and chemical reactions, are challenging and computationally expensive.
MLIPs such as ænet and DeepMD offer promising solutions for bridging the gap between expensive electronic structure methods and efficient classical interatomic potentials. However, incorporating the important effects of valence on chemical bonding remains a challenge in MLIP.
Charge can be represented in a variety of ways, from simple oxidation state labels to continuous wave functions derived from quantum mechanics. The challenge of incorporating charge information into MLIP comes from many factors, such as ambiguity of representation, complexity of interpretation, scarcity of labels, etc.
The content that needs to be rewritten is: CHGNet architecture
CHGNet is pre-trained on energy, force, stress and magnetic moment from the Materials Project Trajectory Dataset (MPtrj). The data set contains more than 10 years of density functional theory calculations on 1.5 million inorganic structures. By explicitly including magnetic moments, CHGNet is able to learn and accurately represent the orbital occupation of electrons, thereby enhancing its ability to describe atomic and electronic degrees of freedom
#MPtrj The distribution of elements in the data set is shown below Shown
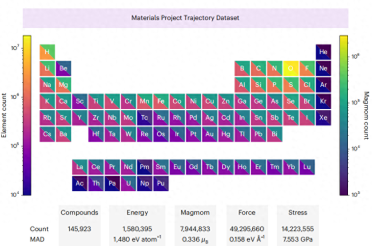
# Rewritten content: Illustration: The distribution of elements in the MPtrj data set. (Source: Paper)
Here, the researchers define charge as an atomic property (atomic charge) that can be inferred by the inclusion of magnetic moments (magmoms). Research shows that by explicitly incorporating site-specific magmoms as charge state constraints into CHGNet, latent space regularization can be enhanced and electronic interactions can be accurately captured
CHGNet is based on GNN, where Figure Vol. Accumulated layers are used to propagate atomic information through a set of nodes {vi} connected by edges {eij}. Translation, rotation and alignment invariance are preserved in GNN. CHGNet takes as input a crystal structure with unknown atomic charges and outputs the corresponding energies, forces, stresses, and magmoms. Charge-decorated structures can be inferred from field magmoms and atomic orbital theory.

The rewritten content is as follows: Illustration: CHGNet model architecture. (Source: paper)
In CHGNet, periodic crystal structures are converted into atomic graphs  by searching for neighboring atoms vj within
by searching for neighboring atoms vj within  of each atom vi in the original unit.
of each atom vi in the original unit.
Different from other GNNs, in which the atomic features updated after t convolutional layers are directly used to predict energy, CHGNet regularizes the node features of t−1 convolutional layers
are directly used to predict energy, CHGNet regularizes the node features of t−1 convolutional layers
 to include information about the magma. Regularized features
to include information about the magma. Regularized features carry rich information about the local ion environment and charge distribution. Therefore, the atomic signature used to predict energy, force, and stress is the charge constrained by information about its charge state. Therefore, CHGNet can provide charge state information using only nuclear positions and atomic identities as inputs, allowing the study of charge distribution in atomic modeling.
carry rich information about the local ion environment and charge distribution. Therefore, the atomic signature used to predict energy, force, and stress is the charge constrained by information about its charge state. Therefore, CHGNet can provide charge state information using only nuclear positions and atomic identities as inputs, allowing the study of charge distribution in atomic modeling. 



Second, although magmom is a good heuristic for atomic charges calculated from spin polarization in ionic systems, it is realized that atomic charge inference for non-magnetic ions can be ambiguous, so Additional domain knowledge is required. Therefore, for ions without magmom, the atom-centered magmom cannot accurately reflect their atomic charge. CHGNet will infer the charge from the environment, similar to other MLIP functions.
can be further improved by combining other charge representation methods Enhanced models such as electron positioning functions, electric polarization and atomic orbital based partitioning. These methods can be used for atomic feature engineering in latent space
CHGNet can implement charge-based atomic simulations and is suitable for large-scale computational simulations to study heterovalent systems, thus expanding the scope of computational chemistry, physics, biology and research opportunities on charge transfer coupling phenomena in materials science
Please click the following link to view the paper: https://www.nature.com/articles/s42256-023-00716-3
The above is the detailed content of Charge-based atomic simulation implementation using pre-trained general purpose neural network CHGNet. For more information, please follow other related articles on the PHP Chinese website!

 Gemma Scope: Google's Microscope for Peering into AI's Thought ProcessApr 17, 2025 am 11:55 AM
Gemma Scope: Google's Microscope for Peering into AI's Thought ProcessApr 17, 2025 am 11:55 AMExploring the Inner Workings of Language Models with Gemma Scope Understanding the complexities of AI language models is a significant challenge. Google's release of Gemma Scope, a comprehensive toolkit, offers researchers a powerful way to delve in
 Who Is a Business Intelligence Analyst and How To Become One?Apr 17, 2025 am 11:44 AM
Who Is a Business Intelligence Analyst and How To Become One?Apr 17, 2025 am 11:44 AMUnlocking Business Success: A Guide to Becoming a Business Intelligence Analyst Imagine transforming raw data into actionable insights that drive organizational growth. This is the power of a Business Intelligence (BI) Analyst – a crucial role in gu
 How to Add a Column in SQL? - Analytics VidhyaApr 17, 2025 am 11:43 AM
How to Add a Column in SQL? - Analytics VidhyaApr 17, 2025 am 11:43 AMSQL's ALTER TABLE Statement: Dynamically Adding Columns to Your Database In data management, SQL's adaptability is crucial. Need to adjust your database structure on the fly? The ALTER TABLE statement is your solution. This guide details adding colu
 Business Analyst vs. Data AnalystApr 17, 2025 am 11:38 AM
Business Analyst vs. Data AnalystApr 17, 2025 am 11:38 AMIntroduction Imagine a bustling office where two professionals collaborate on a critical project. The business analyst focuses on the company's objectives, identifying areas for improvement, and ensuring strategic alignment with market trends. Simu
 What are COUNT and COUNTA in Excel? - Analytics VidhyaApr 17, 2025 am 11:34 AM
What are COUNT and COUNTA in Excel? - Analytics VidhyaApr 17, 2025 am 11:34 AMExcel data counting and analysis: detailed explanation of COUNT and COUNTA functions Accurate data counting and analysis are critical in Excel, especially when working with large data sets. Excel provides a variety of functions to achieve this, with the COUNT and COUNTA functions being key tools for counting the number of cells under different conditions. Although both functions are used to count cells, their design targets are targeted at different data types. Let's dig into the specific details of COUNT and COUNTA functions, highlight their unique features and differences, and learn how to apply them in data analysis. Overview of key points Understand COUNT and COU
 Chrome is Here With AI: Experiencing Something New Everyday!!Apr 17, 2025 am 11:29 AM
Chrome is Here With AI: Experiencing Something New Everyday!!Apr 17, 2025 am 11:29 AMGoogle Chrome's AI Revolution: A Personalized and Efficient Browsing Experience Artificial Intelligence (AI) is rapidly transforming our daily lives, and Google Chrome is leading the charge in the web browsing arena. This article explores the exciti
 AI's Human Side: Wellbeing And The Quadruple Bottom LineApr 17, 2025 am 11:28 AM
AI's Human Side: Wellbeing And The Quadruple Bottom LineApr 17, 2025 am 11:28 AMReimagining Impact: The Quadruple Bottom Line For too long, the conversation has been dominated by a narrow view of AI’s impact, primarily focused on the bottom line of profit. However, a more holistic approach recognizes the interconnectedness of bu
 5 Game-Changing Quantum Computing Use Cases You Should Know AboutApr 17, 2025 am 11:24 AM
5 Game-Changing Quantum Computing Use Cases You Should Know AboutApr 17, 2025 am 11:24 AMThings are moving steadily towards that point. The investment pouring into quantum service providers and startups shows that industry understands its significance. And a growing number of real-world use cases are emerging to demonstrate its value out


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

SublimeText3 English version
Recommended: Win version, supports code prompts!

SecLists
SecLists is the ultimate security tester's companion. It is a collection of various types of lists that are frequently used during security assessments, all in one place. SecLists helps make security testing more efficient and productive by conveniently providing all the lists a security tester might need. List types include usernames, passwords, URLs, fuzzing payloads, sensitive data patterns, web shells, and more. The tester can simply pull this repository onto a new test machine and he will have access to every type of list he needs.

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

VSCode Windows 64-bit Download
A free and powerful IDE editor launched by Microsoft

EditPlus Chinese cracked version
Small size, syntax highlighting, does not support code prompt function

