

 Technology peripheralsAIExploration and application of genome conformation prediction models and high-throughput computational genetic screening methods
Technology peripheralsAIExploration and application of genome conformation prediction models and high-throughput computational genetic screening methodsExploration and application of genome conformation prediction models and high-throughput computational genetic screening methods


Figure 0
#Differences in genome conformation in different types of cells determine the specificity of gene expression , thereby determining the functional differences of different cell types. For a long time, experimental methods for genome conformation detection, from in situ hybridization to high-throughput detection such as Hi-C and micro-C technologies, are usually time-consuming, labor-intensive, costly and have strong technical limitations. These methods greatly limit the widespread application of these experimental techniques in the field of genome conformation research, especially in the study of rare cell types and the need to verify the causal relationship of genome conformation regulation on a large scale. The limitations of these methods have also long restricted new discoveries in the field of three-dimensional genome conformational regulation.
##Picture 1
##January 9, 2023,NYU Grossman School of Medicine’s Aristotelis Tsirigos laboratory and Broad Institute of MIT and Harvard’s Xia Bo laboratory collaborated to publish an article in Nature Biotechnology "Cell type-specific prediction of 3D Chromatin organization enables high-throughput in silico genetic screening》.

In this study, the first authors Tan Jimin and Dr. Xia Bo, doctoral students at New York University School of Medicine, first proposed a new multi-modal machine learning model C.Origami. Predict the chromatin conformation of specific cell types and propose a new high-throughput computational genetic screening (ISGS) method
based on the principles of genetic screening to identify cell type-specific functions Genomic elements help discover new mechanisms of chromatin conformation regulation.
Figure 2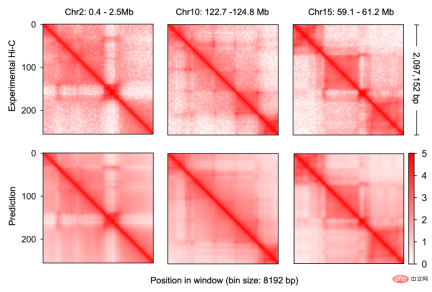
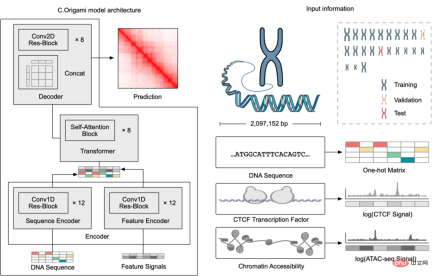
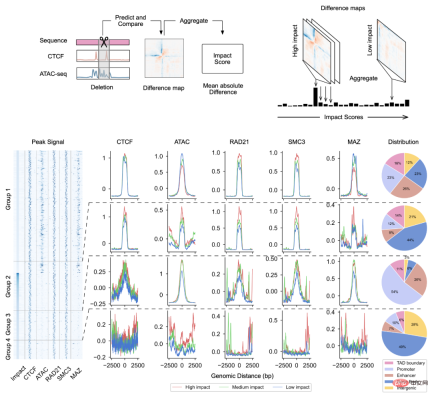
ResearcherFirst A new multi-modal deep learning framework, Origami
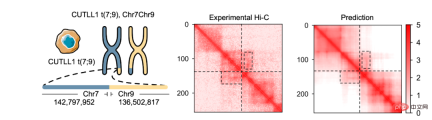
, was constructed for genomic data, enabling it to effectively integrate DNA sequence information and cell-specific functional genomic information to predict new genomic information. Through repeated debugging and model training, the researchers found that integrating DNA sequence, CTCF binding status (CTCF ChIP-seq), and ATAC-seq signals as input information can accurately predict chromatin conformation, and use the two-dimensional Hi-C matrix as Predict the output target (Figure 1-2). Input information was 2 million base pairs of DNA, CTCF ChIP-seq and ATAC-seq. Researchers use Onehot-encoding to encode discrete DNA sequences, while CTCF ChIP-seq and ATAC-seq encode non-discrete features. C.Origami model is divided into three parts, the encoder that processes and compresses DNA and genome information, the Transformer middle layer and the output Hi-C decoder. The encoder consists of a series of 1D ResNet and strided convolution to encode and compress the input information of 2 million base pairs. At the end of the encoder the 2 million length message is compressed to 256 length and used as the input message to the Transformer. Transformer's self-attention mechanism can handle the interdependency between different genomic regions and improve the overall performance of the model. The attention matrix in Transformer can also enhance the interpretability of the model. The researchers converted the attention weight into an "attention score" to measure the model's emphasis on different areas when predicting. Finally, the researchers converted the 1D output of the Transformer module into a 2D contact/adjacency matrix using "outer concatenation", which was used as input information for the Hi-C decoder. The decoder is a Dilated 2D ResNet. The researchers adjusted the dilation factors of different layers so that the receptive field at each pixel position of the final layer can cover all input information. This model for predicting chromatin conformation is called C.Origami. Researchers call C.Origami the first multimodal deep learning model in genomics. Due to its multimodal nature, C.Origami is able to accurately predict (de novo prediction) chromatin conformation in new cell types that have never been exposed before. For example, a model trained on IMR-90 cells (lung fibroblasts) was able to accurately predict specific chromatin conformations in GM12878 cells (B lymphocytes) (Figure 3). Figure 3 structural variant --- - Such as chromosomal translocations - are very common in tumors and often alter chromatin interaction patterns, which may affect the expression of oncogenes or tumor suppressor genes. Studying the effects of these structural variations on chromatin conformation and gene expression is important for understanding the mechanisms of tumor occurrence and progression. This type of research usually requires the use of experiments such as 4C-seq or Hi-C to analyze the chromatin conformation of structural variation sites, but is often limited by resources and time and is difficult to conduct on a large scale. In this study, C. Origami can model variations in DNA sequences in input variables and then predict new chromatin interactions in the mutated cancer genome. Previous studies found that the T-cell acute lymphoblastic leukemia (T-ALL) cell model CUTLL1 has a chr7-chr9 chromosomal translocation (Figure 4). By computationally simulating chromosomal translocation variants, C. Origami accurately predicted the new TAD structure at the variant site and detected a ‘chromatin stripe’ structure extending from chr9 to chr7 (Figure 4). Figure 4 In view of the accurate prediction effect of C.Origami, Inspired by the principle of reverse genetic screening, researchers proposed a new high-throughput computational genetic screening (ISGS) method to systematically identify cell type-specific functional genomic elements and help discover new ones. Staining regulatory molecules (Figure 5). The researchers developed a framework for computational genetic screening ISGS based on the C. Origami model for the systematic identification of cis-regulatory elements required for chromatin conformation. Using genome-wide 1kb resolution ISGS, the authors isolated cis-regulatory elements (∼1% of the genome) that have important effects on chromatin conformation. These chromatin conformation regulatory sequences exhibit differential dependence on CTCF binding and ATAC-seq signals (Fig. 5 ). Figure 5 The ISGS framework enables high-throughput screening of cell- or disease-specific chromatin conformations. The researchers performed ISGS in CUTLL1, Jurkat and normal T cells and found that a cis-regulatory element (CHD4-insu) near the CHD4 gene was specifically lost in T-ALL cells. Screening results indicate that the insulating loss of CHD4-insu in T-ALL cells may enable the CHD4 gene to establish new chromatin interactions, thereby upregulating CHD4 expression and promoting leukemia cell proliferation. ISGS can also be used to systematically discover novel trans-acting factors that regulate chromatin conformation. Through enrichment analysis of important cell type-specific regulatory sequences and transcription factor binding sites, the researchers identified regulatory factors that contribute to cell type-specific genome conformation. Interestingly, previous studies found that MAZ may regulate chromatin conformation together with CTCF. Through ISGS and transcription factor enrichment analysis, the authors found that MAZ is greatly enriched in open chromatin regions, while showing only weak binding in non-open chromatin regions where CTCF binds. This result suggests that MAZ may regulate genome conformation independently of CTCF. Researchers see great potential in multimodal machine learning models that combine DNA sequence and chromatin information in chromatin structure prediction. The underlying multimodal architecture of the model, Origami, can be extended to the application of other genomics data, such as epigenetic modifications, gene expression, functional screening of mutations, etc. Researchers predict that future genomics research will shift more towards the use of deep learning models as tools for primary computational genetic screening, supplemented by a new generation of high-throughput research methods validated by biological experiments. In this study, Tan Jimin, a doctoral candidate at New York University School of Medicine, is the first author, and Dr. Aristotelis Tsirigos and Dr. Xia Bo are the co-corresponding authors. This research began with the brainstorming of Xia Bo and Tan Jimin during the epidemic lockdown in October 2020. After two and a half years of improvement and polishing, it was officially published in Nature Biotechnology in January 2023. The code and training data of this project have been open sourced on GitHub and Zenodo, and are equipped with Google Colab for functional demonstration. Project address: https://github.com/tanjimin/C.Origami Dr. Xia Bo’s laboratory (Broad Institute of MIT and Harvard) homepage: www.boxialab.org Dr. Xia Bo is committed to analyzing the core mechanism and regulation of the three-dimensional conformation of the genome. Its biological significance for human disease, development and evolution. Xia Bo's laboratory welcomes like-minded postdocs to join the team. Tsirigos Lab (New York University Grossman School of Medicine) Home page: http://www.tsirigos.com Tsirigos Lab’s main page Research interests include the application of chromatin, epigenetics and machine learning in precision medicine. 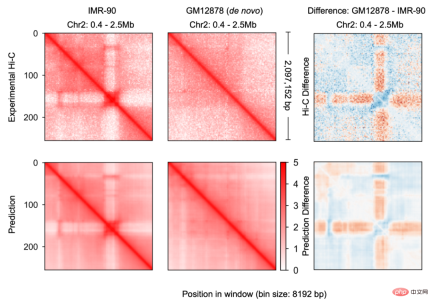
Corresponding author
The above is the detailed content of Exploration and application of genome conformation prediction models and high-throughput computational genetic screening methods. For more information, please follow other related articles on the PHP Chinese website!

 Let's Dance: Structured Movement To Fine-Tune Our Human Neural NetsApr 27, 2025 am 11:09 AM
Let's Dance: Structured Movement To Fine-Tune Our Human Neural NetsApr 27, 2025 am 11:09 AMScientists have extensively studied human and simpler neural networks (like those in C. elegans) to understand their functionality. However, a crucial question arises: how do we adapt our own neural networks to work effectively alongside novel AI s
 New Google Leak Reveals Subscription Changes For Gemini AIApr 27, 2025 am 11:08 AM
New Google Leak Reveals Subscription Changes For Gemini AIApr 27, 2025 am 11:08 AMGoogle's Gemini Advanced: New Subscription Tiers on the Horizon Currently, accessing Gemini Advanced requires a $19.99/month Google One AI Premium plan. However, an Android Authority report hints at upcoming changes. Code within the latest Google P
 How Data Analytics Acceleration Is Solving AI's Hidden BottleneckApr 27, 2025 am 11:07 AM
How Data Analytics Acceleration Is Solving AI's Hidden BottleneckApr 27, 2025 am 11:07 AMDespite the hype surrounding advanced AI capabilities, a significant challenge lurks within enterprise AI deployments: data processing bottlenecks. While CEOs celebrate AI advancements, engineers grapple with slow query times, overloaded pipelines, a
 MarkItDown MCP Can Convert Any Document into Markdowns!Apr 27, 2025 am 09:47 AM
MarkItDown MCP Can Convert Any Document into Markdowns!Apr 27, 2025 am 09:47 AMHandling documents is no longer just about opening files in your AI projects, it’s about transforming chaos into clarity. Docs such as PDFs, PowerPoints, and Word flood our workflows in every shape and size. Retrieving structured
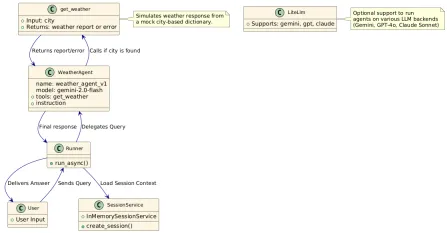 How to Use Google ADK for Building Agents? - Analytics VidhyaApr 27, 2025 am 09:42 AM
How to Use Google ADK for Building Agents? - Analytics VidhyaApr 27, 2025 am 09:42 AMHarness the power of Google's Agent Development Kit (ADK) to create intelligent agents with real-world capabilities! This tutorial guides you through building conversational agents using ADK, supporting various language models like Gemini and GPT. W
 Use of SLM over LLM for Effective Problem Solving - Analytics VidhyaApr 27, 2025 am 09:27 AM
Use of SLM over LLM for Effective Problem Solving - Analytics VidhyaApr 27, 2025 am 09:27 AMsummary: Small Language Model (SLM) is designed for efficiency. They are better than the Large Language Model (LLM) in resource-deficient, real-time and privacy-sensitive environments. Best for focus-based tasks, especially where domain specificity, controllability, and interpretability are more important than general knowledge or creativity. SLMs are not a replacement for LLMs, but they are ideal when precision, speed and cost-effectiveness are critical. Technology helps us achieve more with fewer resources. It has always been a promoter, not a driver. From the steam engine era to the Internet bubble era, the power of technology lies in the extent to which it helps us solve problems. Artificial intelligence (AI) and more recently generative AI are no exception
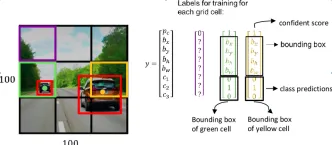 How to Use Google Gemini Models for Computer Vision Tasks? - Analytics VidhyaApr 27, 2025 am 09:26 AM
How to Use Google Gemini Models for Computer Vision Tasks? - Analytics VidhyaApr 27, 2025 am 09:26 AMHarness the Power of Google Gemini for Computer Vision: A Comprehensive Guide Google Gemini, a leading AI chatbot, extends its capabilities beyond conversation to encompass powerful computer vision functionalities. This guide details how to utilize
 Gemini 2.0 Flash vs o4-mini: Can Google Do Better Than OpenAI?Apr 27, 2025 am 09:20 AM
Gemini 2.0 Flash vs o4-mini: Can Google Do Better Than OpenAI?Apr 27, 2025 am 09:20 AMThe AI landscape of 2025 is electrifying with the arrival of Google's Gemini 2.0 Flash and OpenAI's o4-mini. These cutting-edge models, launched weeks apart, boast comparable advanced features and impressive benchmark scores. This in-depth compariso


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

SublimeText3 English version
Recommended: Win version, supports code prompts!

SecLists
SecLists is the ultimate security tester's companion. It is a collection of various types of lists that are frequently used during security assessments, all in one place. SecLists helps make security testing more efficient and productive by conveniently providing all the lists a security tester might need. List types include usernames, passwords, URLs, fuzzing payloads, sensitive data patterns, web shells, and more. The tester can simply pull this repository onto a new test machine and he will have access to every type of list he needs.

Dreamweaver Mac version
Visual web development tools

Notepad++7.3.1
Easy-to-use and free code editor

PhpStorm Mac version
The latest (2018.2.1) professional PHP integrated development tool

