
 Technology peripheralsAIA Chinese team successfully developed AI to predict suitable drugs for cancer patients, and the results were published in the Nature sub-journal
Technology peripheralsAIA Chinese team successfully developed AI to predict suitable drugs for cancer patients, and the results were published in the Nature sub-journalA Chinese team successfully developed AI to predict suitable drugs for cancer patients, and the results were published in the Nature sub-journal

With just one AI, the clinical responses of 9,808 cancer patients to drugs can be fully predicted.
And the results are consistent with clinical observations.
This is the latest result CODE-AE (context-aware deconfounding autoencoder) brought by Lei Xie's team at the City University of New York.

It proposes a novel contextual autoencoding model that can predict the specific responses of different patients to drugs.
This will have a significant impact on new drug development and clinical trials.
You must know that under the traditional model, it takes nearly 10 years to develop, test, and fully market a new drug, and the funds consumed are unprecedentedly huge, easily reaching 1 billion U.S. dollars.
The cycle is so long because the reaction of new drugs in the human body is difficult to predict, and repeated trials are often required for testing.
If AI can use data to make predictions, it will significantly shorten the time to market for new drugs and reduce costs.
Currently, this research has been published in the Nature sub-journal "Nature Machine Intelligence".
Simply put, CODE-AE uses data from in vitro cell validation of new drugs to predict the response of the drug in the human body.
This avoids the dependence of AI model training on patient clinical data.
The biggest reason why AI has not been very effective in clinical response prediction in the past is that it is too difficult to collect massive and continuous clinical response data.
From a mechanism perspective, researchers divide drug biomarkers into source domain and target domain.
The source domain represents a different domain than the test sample, but has rich supervision information, which can be understood as in vitro cell verification data.
The target domain is the domain where the test sample is located. It has no labels or only a small number of labels, that is, patient data.

Map data features from different fields to the same feature space so that their distances in this space are as close as possible.
So the objective function trained on the source domain in the feature space can be transferred to the target domain to improve the accuracy in the target domain.
In the context of this research, the source domain and the target domain are both data characteristics of drug biomarkers, that is, data characteristics of drug targets.
Looking specifically at the model framework, it is mainly divided into three parts: pre-training, fine-tuning and inference.
Pre-training mainly uses self-supervised learning to build a feature encoding module to map the unlabeled gene expression profiles of in vitro cell data and patient data into the embedding space. In this way, some confounding factors can be eliminated and the latent distribution of the two data can be consistent to eliminate systematic bias.
The fine-tuning stage is to add a supervised model on the basis of pre-training and use labeled in vitro cell data for training.
Finally, in the inference stage, the patients obtained from pre-training are first disambiguated and embedded, and then the tuned model is used to predict the patient's response to the drug.

In this mode, CODE-AE has two characteristics.
First, it can extract common biological signals and private representations in incoherent samples, thereby eliminating interference caused by different data patterns.
Second, after separating the drug response signal and confounding factors, local alignment can also be achieved.
To summarize, CODE-AE can be understood as the process of selecting unique features in the incoherent data pattern embedding space of labeled and unlabeled data.
To demonstrate the effectiveness of the model, the researchers predicted the drug suitability of 9,808 cancer patients.
If the site results predicted by the model for the patient's condition are related to the drug target he uses, it proves that the prediction is correct.
The researchers then divided the patients into 100 clusters and the 59 drugs into 30 clusters.
Through this analysis method, patients with similar drug response profiles can be grouped together.
Here, we take the clustering of patients with lung squamous cell carcinoma (LSCC) and non-small cell lung cancer (NSCLC) as an example.
Among the 59 drugs, the most sensitive drugs for LSCC are gefitinib, AICAR and gemcitabine.
The targets of gefitinib and AICAR are both epidermal growth factor receptors (EGFR), and gemcitabine is often used to treat non-small cell lung cancer without EGFR mutations.
The paper stated that, consistent with the action modes of these drugs, CODE-AE found that patients using gefitinib and AICAR had similar drug response profiles.
In other words, CODE-AE has discovered the correct target for patient treatment, that is, it can predict applicable drugs.

The above research team is from the City University of New York.
The corresponding author is Lei Xie, who graduated from the University of Science and Technology of China in polymer physics.
He graduated with a master's degree in computer science from Rutgers University; his doctorate was also from Rutgers University, but with a degree in chemistry.

It is understood that the next step of the research team will be to develop the CODE-AE prediction function for the concentration and metabolism of the clinical response of new drugs.
The researchers said that the AI model may also be adapted to predict the side effects of drugs on the human body.
It is worth mentioning that the Nature sub-journal "Nature Machine Intelligence" specializes in interdisciplinary applied research in artificial intelligence and life sciences, and the average number of papers included every year is about 60.
Paper address: https://www.nature.com/articles/s42256-022-00541-0
Reference link: https://phys.org/news/2022-10 -ai-accurately-human-response-drug.html
The above is the detailed content of A Chinese team successfully developed AI to predict suitable drugs for cancer patients, and the results were published in the Nature sub-journal. For more information, please follow other related articles on the PHP Chinese website!

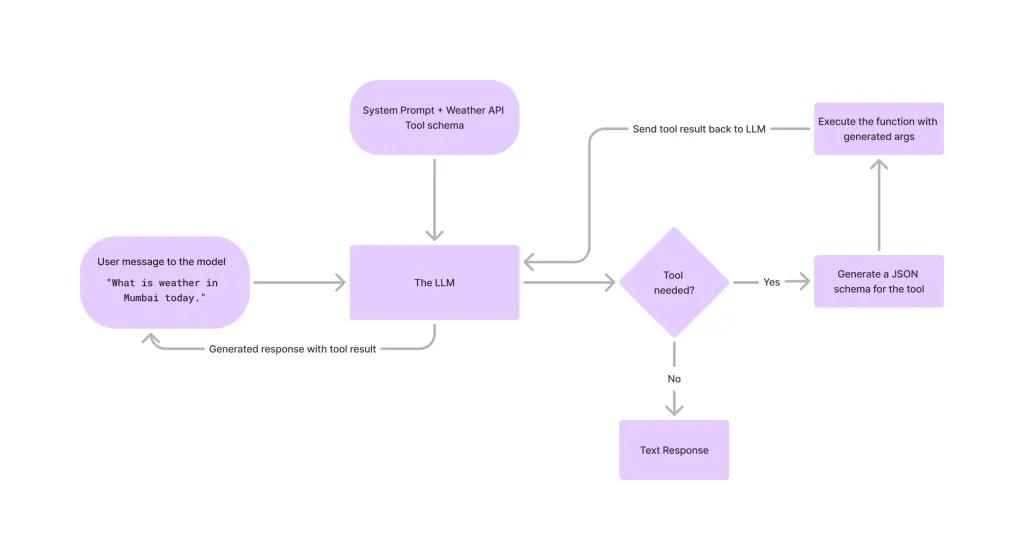 Tool Calling in LLMsApr 14, 2025 am 11:28 AM
Tool Calling in LLMsApr 14, 2025 am 11:28 AMLarge language models (LLMs) have surged in popularity, with the tool-calling feature dramatically expanding their capabilities beyond simple text generation. Now, LLMs can handle complex automation tasks such as dynamic UI creation and autonomous a
 How ADHD Games, Health Tools & AI Chatbots Are Transforming Global HealthApr 14, 2025 am 11:27 AM
How ADHD Games, Health Tools & AI Chatbots Are Transforming Global HealthApr 14, 2025 am 11:27 AMCan a video game ease anxiety, build focus, or support a child with ADHD? As healthcare challenges surge globally — especially among youth — innovators are turning to an unlikely tool: video games. Now one of the world’s largest entertainment indus
 UN Input On AI: Winners, Losers, And OpportunitiesApr 14, 2025 am 11:25 AM
UN Input On AI: Winners, Losers, And OpportunitiesApr 14, 2025 am 11:25 AM“History has shown that while technological progress drives economic growth, it does not on its own ensure equitable income distribution or promote inclusive human development,” writes Rebeca Grynspan, Secretary-General of UNCTAD, in the preamble.
 Learning Negotiation Skills Via Generative AIApr 14, 2025 am 11:23 AM
Learning Negotiation Skills Via Generative AIApr 14, 2025 am 11:23 AMEasy-peasy, use generative AI as your negotiation tutor and sparring partner. Let’s talk about it. This analysis of an innovative AI breakthrough is part of my ongoing Forbes column coverage on the latest in AI, including identifying and explaining
 TED Reveals From OpenAI, Google, Meta Heads To Court, Selfie With MyselfApr 14, 2025 am 11:22 AM
TED Reveals From OpenAI, Google, Meta Heads To Court, Selfie With MyselfApr 14, 2025 am 11:22 AMThe TED2025 Conference, held in Vancouver, wrapped its 36th edition yesterday, April 11. It featured 80 speakers from more than 60 countries, including Sam Altman, Eric Schmidt, and Palmer Luckey. TED’s theme, “humanity reimagined,” was tailor made
 Joseph Stiglitz Warns Of The Looming Inequality Amid AI Monopoly PowerApr 14, 2025 am 11:21 AM
Joseph Stiglitz Warns Of The Looming Inequality Amid AI Monopoly PowerApr 14, 2025 am 11:21 AMJoseph Stiglitz is renowned economist and recipient of the Nobel Prize in Economics in 2001. Stiglitz posits that AI can worsen existing inequalities and consolidated power in the hands of a few dominant corporations, ultimately undermining economic
 What is Graph Database?Apr 14, 2025 am 11:19 AM
What is Graph Database?Apr 14, 2025 am 11:19 AMGraph Databases: Revolutionizing Data Management Through Relationships As data expands and its characteristics evolve across various fields, graph databases are emerging as transformative solutions for managing interconnected data. Unlike traditional
 LLM Routing: Strategies, Techniques, and Python ImplementationApr 14, 2025 am 11:14 AM
LLM Routing: Strategies, Techniques, and Python ImplementationApr 14, 2025 am 11:14 AMLarge Language Model (LLM) Routing: Optimizing Performance Through Intelligent Task Distribution The rapidly evolving landscape of LLMs presents a diverse range of models, each with unique strengths and weaknesses. Some excel at creative content gen


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

PhpStorm Mac version
The latest (2018.2.1) professional PHP integrated development tool

SublimeText3 English version
Recommended: Win version, supports code prompts!

WebStorm Mac version
Useful JavaScript development tools

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

Zend Studio 13.0.1
Powerful PHP integrated development environment

