

 Technology peripheralsAIMeta AI opens 600 million+ metagenomic protein structure maps, and 15 billion language models were completed in two weeks
Technology peripheralsAIMeta AI opens 600 million+ metagenomic protein structure maps, and 15 billion language models were completed in two weeks
This year, DeepMind published the predicted structures of approximately 220 million proteins, which covers almost all proteins of known organisms in the DNA database. Now another tech giant, Meta, is filling another void, that of microbes.
Simply put, Meta uses AI technology to predict the structures of approximately 600 million proteins from bacteria and other as-yet-uncharacterized microorganisms. Team leader Alexander Rives said: "These proteins are the structures that we know the least about, and they are very mysterious proteins. I think these findings provide the potential for a deeper understanding of biology."
Typically, language models are trained on large amounts of text. Meta To apply language models to proteins, Rives and colleagues took as input known protein sequences, which are composed of 20 amino acids represented by different letters. The network then learned to automatically complete proteins while masking a certain proportion of amino acids.
Meta named this network ESMFold. Although ESMFold's prediction accuracy is not as good as AlphaFold's, it is about 60 times faster than AlphaFold in predicting structures. This speed means protein structure predictions can be scaled up to larger databases.

- Paper address: https://www.biorxiv.org/content/10.1101/2022.07.20.500902v2
- Project address: https://github.com/facebookresearch/esm
Now, As a test, Meta decided to apply their model to a database of metagenomic DNA, all sourced from the environment, including soil, seawater, the human gut, skin and other microbial habitats. Meta AI announced the launch of the ESM Metagenomic Atlas containing more than 600 million proteins, which is the first comprehensive view of the "dark matter" of the protein universe. It is also the largest database of high-resolution predicted structures, 3 times larger than any existing protein structure database, and the first to provide comprehensive, large-scale coverage of metagenomic proteins.
In total, the Meta team predicted more than 617 million protein structures in just two weeks. Rives said the predictions are free and available to anyone, just like the underlying code of the model.
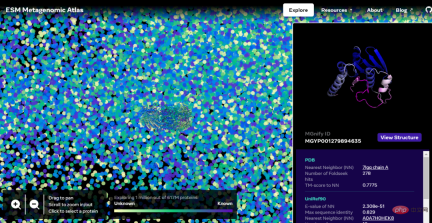
##Interactive version address: https://esmatlas.com/explore?at=1,1,21.999999344348925
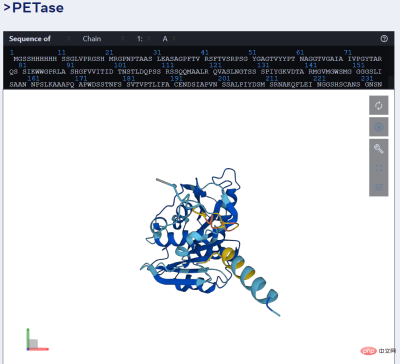
For example, the picture below shows the prediction of PET enzyme by ESMFold.
As we all know, proteins are complex and dynamic molecules encoded by genes and are mainly responsible for the basic processes of life. Proteins have amazing roles in biology. For example, the rods and cones in the human eye can sense light, so we can see the outside world; the molecular sensors that form the basis of hearing and touch; the complex molecules in plants that convert light energy into chemical energy; the molecules that drive microorganisms and The "motors" that make human muscles move; the enzymes that break down plastic; the antibodies that protect us from disease, etc. are all proteins.
In 1998, Jo Handelsman from the Department of Plant Pathology at the University of Wisconsin first proposed the concept of metagenomics. To a certain extent, it can be regarded as the idea of research and analysis of a single genome, and the English word for macro is meta-, which is also translated as meta.
Metagenomics reveals billions of protein sequences that are new to science and cataloged for the first time by NCBI, the European Bioinformatics Institute and in large databases compiled by public projects such as the Joint Genome Institute.
Novel protein folding method developed by Meta AI that leverages large language models to create the first comprehensive view of protein structure in metagenomic databases (with hundreds of millions of proteins). Meta found that language models can predict the atomic-level three-dimensional structure of proteins 60 times faster than existing SOTA protein structure prediction methods. This advance will help accelerate a new era of protein structure understanding, making it possible for the first time to understand the structures of the billions of proteins being cataloged by genetic sequencing technology.
Unlocking the hidden world of nature: the first comprehensive view of metagenomic structural space
We know that advances in genetic sequencing have enabled the analysis of billions of metagenomic protein sequences Cataloging becomes possible. But experimentally determining the 3D structure of billions of proteins goes well beyond the scope of time-intensive laboratory techniques such as X-ray crystallography, which can take weeks or even years to detect a single protein. Computational approaches can provide insights into metagenomics proteins that are not possible using experimental techniques.
ESM Metagenomic Maps will enable scientists to search and analyze the structure of metagenomic proteins on a scale of hundreds of millions of proteins. This can help identify previously uncharacterized structures, search for distant evolutionary relationships, and discover new proteins that could be used in medicine and other applications.
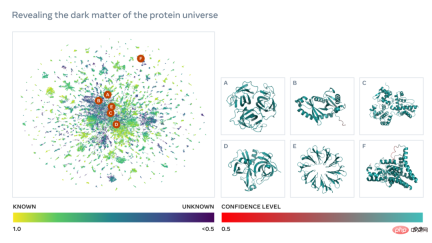
Below is a map containing tens of thousands of high-confidence predictions, showing similarities to proteins with currently known structures. And, for the first time, the image shows a much larger region of protein structure space that was completely unknown.
Learn to read biological language
As shown in the figure below, the ESM-2 language model has been trained to predict the evolution process Amino acids masked by sequence. Meta AI found that, as a result of training, information about protein structure emerged in the model’s internal state. This is really surprising since the model was only trained on sequences.

Just like the text of a paper or letter, proteins can be written as sequences of characters. Each character corresponds to one of 20 standard chemical elements (amino acids), each with different properties and which are the building blocks of proteins. These building blocks can be put together in astronomically different ways, for example for a protein consisting of 200 amino acids, there are 20^200 possible sequences, which is more than the number of atoms in the visible universe. Each sequence folds into a 3D shape (but not all sequences fold into a coherent structure, many fold into a disordered form), and it is this shape that largely determines the biological function of the protein.
Learning to read the biological language brings great challenges. While both protein sequences and text passages can be written as characters, there are deep and fundamental differences between them. A protein sequence describes the chemical structure of a molecule that folds into complex 3D shapes according to the laws of physics.
Protein sequences contain statistical patterns that convey information about protein folding structure. For example, if two positions in a protein co-evolve, or in other words, if a certain amino acid occurs at one position that usually pairs with a certain amino acid at the other position, this may mean that the two positions are in the folded structure interaction. This is similar to two pieces of a jigsaw puzzle, where evolution must choose the amino acids that fit together in a folded structure. This in turn means that we can often infer the structure of a protein by observing patterns in its sequence.
ESM uses AI to learn to read these patterns. In 2019, Meta AI provided evidence that language models learned the properties of proteins, such as their structure and function. Through a form of self-supervised learning called masked language modeling, Meta AI trained a language model on the sequences of millions of natural proteins. Using this method, the model must correctly fill in the blanks in the text paragraph, such as "To _ or not to , that is the _____".
After that, Meta AI trained a language model to fill in the gaps in the protein sequence. They found that information about protein structure and function emerged during this training. In 2020, Meta released a SOTA protein language model, ESM1b, for a variety of applications, including helping scientists predict the evolution of COVID-19 and discover the genetic causes of the disease.
Now, Meta AI has extended this approach to create the next generation protein language model ESM-2, which at 15 billion parameters is the largest protein language model to date. They found that when the model parameters were scaled up from 8 million to 15 billion, information emerged in the internal representation, enabling 3D structure predictions at atomic resolution.
Achieve orders of magnitude acceleration in protein folding
In the figure below, as the model is enlarged, a high-resolution protein structure appears. At the same time, as the model is scaled, new details appear in atomic-resolution images of the protein structure.

Using current SOTA computational tools, predicting the structure of hundreds of millions of protein sequences in a realistic time frame would take years, even using major research institutions The same goes for resources. Therefore, to make predictions at the metagenomic scale, a breakthrough in prediction speed is crucial.
Meta AI finds that using language models of protein sequences significantly speeds up structure predictions, by up to 60 times. This is sufficient to make predictions on an entire metagenomic database in just a few weeks and can be scaled to much larger databases than our currently published ones. In fact, this new structure prediction capability was able to predict the sequences of more than 600 million metagenome proteins in just two weeks on a cluster of approximately 2,000 GPUs.
Additionally, current SOTA structure prediction methods require searching large protein databases to identify relevant sequences. These methods require essentially a whole set of evolutionarily related sequences as input so that they can extract structure-related patterns. Meta AI's ESM-2 language model learns these evolutionary patterns during its training on protein sequences, enabling high-resolution predictions of 3D structures directly from protein sequences.
The figure below shows protein folding using the ESM-2 language model. The arrows from left to right show the flow of information in the network from the language model to the folding trunk to the structural module, and finally outputs 3D coordinates and confidence.
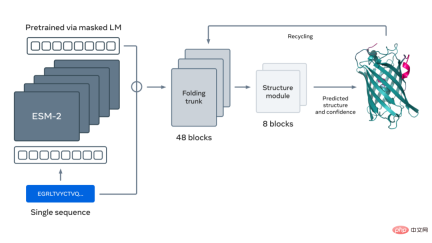
Please refer to the original article for more details.
Blog link: https://ai.facebook.com/blog/protein-folding-esmfold-metagenomics/
The above is the detailed content of Meta AI opens 600 million+ metagenomic protein structure maps, and 15 billion language models were completed in two weeks. For more information, please follow other related articles on the PHP Chinese website!

 ai合并图层的快捷键是什么Jan 07, 2021 am 10:59 AM
ai合并图层的快捷键是什么Jan 07, 2021 am 10:59 AMai合并图层的快捷键是“Ctrl+Shift+E”,它的作用是把目前所有处在显示状态的图层合并,在隐藏状态的图层则不作变动。也可以选中要合并的图层,在菜单栏中依次点击“窗口”-“路径查找器”,点击“合并”按钮。
 ai橡皮擦擦不掉东西怎么办Jan 13, 2021 am 10:23 AM
ai橡皮擦擦不掉东西怎么办Jan 13, 2021 am 10:23 AMai橡皮擦擦不掉东西是因为AI是矢量图软件,用橡皮擦不能擦位图的,其解决办法就是用蒙板工具以及钢笔勾好路径再建立蒙板即可实现擦掉东西。
 谷歌超强AI超算碾压英伟达A100!TPU v4性能提升10倍,细节首次公开Apr 07, 2023 pm 02:54 PM
谷歌超强AI超算碾压英伟达A100!TPU v4性能提升10倍,细节首次公开Apr 07, 2023 pm 02:54 PM虽然谷歌早在2020年,就在自家的数据中心上部署了当时最强的AI芯片——TPU v4。但直到今年的4月4日,谷歌才首次公布了这台AI超算的技术细节。论文地址:https://arxiv.org/abs/2304.01433相比于TPU v3,TPU v4的性能要高出2.1倍,而在整合4096个芯片之后,超算的性能更是提升了10倍。另外,谷歌还声称,自家芯片要比英伟达A100更快、更节能。与A100对打,速度快1.7倍论文中,谷歌表示,对于规模相当的系统,TPU v4可以提供比英伟达A100强1.
 ai可以转成psd格式吗Feb 22, 2023 pm 05:56 PM
ai可以转成psd格式吗Feb 22, 2023 pm 05:56 PMai可以转成psd格式。转换方法:1、打开Adobe Illustrator软件,依次点击顶部菜单栏的“文件”-“打开”,选择所需的ai文件;2、点击右侧功能面板中的“图层”,点击三杠图标,在弹出的选项中选择“释放到图层(顺序)”;3、依次点击顶部菜单栏的“文件”-“导出”-“导出为”;4、在弹出的“导出”对话框中,将“保存类型”设置为“PSD格式”,点击“导出”即可;
 ai顶部属性栏不见了怎么办Feb 22, 2023 pm 05:27 PM
ai顶部属性栏不见了怎么办Feb 22, 2023 pm 05:27 PMai顶部属性栏不见了的解决办法:1、开启Ai新建画布,进入绘图页面;2、在Ai顶部菜单栏中点击“窗口”;3、在系统弹出的窗口菜单页面中点击“控制”,然后开启“控制”窗口即可显示出属性栏。
 GPT-4的研究路径没有前途?Yann LeCun给自回归判了死刑Apr 04, 2023 am 11:55 AM
GPT-4的研究路径没有前途?Yann LeCun给自回归判了死刑Apr 04, 2023 am 11:55 AMYann LeCun 这个观点的确有些大胆。 「从现在起 5 年内,没有哪个头脑正常的人会使用自回归模型。」最近,图灵奖得主 Yann LeCun 给一场辩论做了个特别的开场。而他口中的自回归,正是当前爆红的 GPT 家族模型所依赖的学习范式。当然,被 Yann LeCun 指出问题的不只是自回归模型。在他看来,当前整个的机器学习领域都面临巨大挑战。这场辩论的主题为「Do large language models need sensory grounding for meaning and u
 强化学习再登Nature封面,自动驾驶安全验证新范式大幅减少测试里程Mar 31, 2023 pm 10:38 PM
强化学习再登Nature封面,自动驾驶安全验证新范式大幅减少测试里程Mar 31, 2023 pm 10:38 PM引入密集强化学习,用 AI 验证 AI。 自动驾驶汽车 (AV) 技术的快速发展,使得我们正处于交通革命的风口浪尖,其规模是自一个世纪前汽车问世以来从未见过的。自动驾驶技术具有显着提高交通安全性、机动性和可持续性的潜力,因此引起了工业界、政府机构、专业组织和学术机构的共同关注。过去 20 年里,自动驾驶汽车的发展取得了长足的进步,尤其是随着深度学习的出现更是如此。到 2015 年,开始有公司宣布他们将在 2020 之前量产 AV。不过到目前为止,并且没有 level 4 级别的 AV 可以在市场
 ai移动不了东西了怎么办Mar 07, 2023 am 10:03 AM
ai移动不了东西了怎么办Mar 07, 2023 am 10:03 AMai移动不了东西的解决办法:1、打开ai软件,打开空白文档;2、选择矩形工具,在文档中绘制矩形;3、点击选择工具,移动文档中的矩形;4、点击图层按钮,弹出图层面板对话框,解锁图层;5、点击选择工具,移动矩形即可。


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

Dreamweaver CS6
Visual web development tools

DVWA
Damn Vulnerable Web App (DVWA) is a PHP/MySQL web application that is very vulnerable. Its main goals are to be an aid for security professionals to test their skills and tools in a legal environment, to help web developers better understand the process of securing web applications, and to help teachers/students teach/learn in a classroom environment Web application security. The goal of DVWA is to practice some of the most common web vulnerabilities through a simple and straightforward interface, with varying degrees of difficulty. Please note that this software

WebStorm Mac version
Useful JavaScript development tools

Atom editor mac version download
The most popular open source editor

MinGW - Minimalist GNU for Windows
This project is in the process of being migrated to osdn.net/projects/mingw, you can continue to follow us there. MinGW: A native Windows port of the GNU Compiler Collection (GCC), freely distributable import libraries and header files for building native Windows applications; includes extensions to the MSVC runtime to support C99 functionality. All MinGW software can run on 64-bit Windows platforms.

