

 Technology peripheralsAIEpic “Crossover” Between AlphaFold 3 and GPT-4o’s Knowledge of Protein Data Bank Entries
Technology peripheralsAIEpic “Crossover” Between AlphaFold 3 and GPT-4o’s Knowledge of Protein Data Bank EntriesEpic “Crossover” Between AlphaFold 3 and GPT-4o’s Knowledge of Protein Data Bank Entries

If you are into bioinformatics and data analysis for biology, you will find this article quite inspiring right away.
More broadly for AI scientists, they will find here ways to probe an LLM by pushing it to hallucinate, and then finding ways to overcome this limitation.
Introduction
The Protein Data Bank (PDB) serves as a comprehensive repository for three-dimensional structural data of biological macromolecules, providing invaluable insights into the molecular underpinnings of biological processes. Its mere existence is what allowed AI models like AlphaFold to be developed!
Here are all my peer-reviewed and blog articles on protein modeling, CASP, and AlphaFold 2
"Sparks of Chemical Intuition"—and Gross Limitations!—in AlphaFold 3
Efficiently browsing and searching entries in the PDB is essential for modern work in biology; however, despite a quite complete search engine, several questions are hard to pose. But it turns out that, as I found and report here, we can now interrogate the PDB with natural language requests because, as you saw in this title’s article, GPT-4o knows the Protein Data Bank!
Into it
From some tests I did out of curiosity, I found out that OpenAI clearly has included PDB content (or content that includes PDB information, I’ll discuss this later on) in the training of some of its large language models (LLMs), at least in the training of GPT-4o. After having found this out, I set myself to play with this and then coupled some ideas that came up from my chats with GPT-4o with structure determination tests using AlphaFold 3.
What I envision based on my results is that with the advancements in Artificial Intelligence, particularly in natural language processing models, researchers can now more easily search and thus utilize pertinent structural information from the PDB. The capability to query the PDB with natural language requests is quite powerful as you will see in the many examples I present, allowing for an in-depth exploration of biomolecular structures, complexes and assemblies, including not just folded proteins but also nucleic acids, disordered proteins, and small molecules, all with direct links to experimentally validated structural data in the form of associated PDB entries.
More specifically, through many examples I will demonstrate how GPT-4o can serve to identify relevant PDB entries associated with a complex search term that might not work well if you just enter it in the regular search box available at the PDB sites. My examples cover some quite relevant interactions for biology and pharma, such as Glivec (imatinib, an anticancer drug) bound to various kinase targets, intrinsically disordered regions (IDRs) engaging with binding partners, and post-translational modifications (PTMs) influencing interactions. In some of the examples I further explore the potential of using AlphaFold 3 to model these interactions in space, effectively providing a bridge between LLM-based PDB retrieval and 3D visualization.
In what follows, I phrase my investigation as questions asked to GPT-4o followed by its raw answers and in some cases pictures I prepared to illustrate some downstream analyses I did on selected examples.
Exploration 1: Do you know PDB codes for some example of glivec (imatinib) bound to a kinase?
I asked ChatGPT-4o "Do you know PDB codes for some example of glivec (imatinib) bound to a kinase?" and…
ChatGPT said:
Yes, Glivec (imatinib) is a well-known tyrosine kinase inhibitor, and several structures in the PDB show it bound to its target kinases. Here are some key examples:
- Abl Kinase Bound to Imatinib
PDB Code: 1IEP
Description: This is one of the earliest structures showing imatinib bound to the Abl kinase, the target in chronic myelogenous leukemia (CML). Imatinib binds to the inactive conformation of Abl, stabilizing it and preventing kinase activation.
Here’s my check – I did this for all examples provided by ChatGPT-4o, but I show only some. You see here that PDB 1IEP is a kinase domain complexed with STI-571, which corresponds to imatinib in PDB’s ligand specification:

ChatGPT-4o then continued with more examples:
- c-Kit Kinase Bound to Imatinib
PDB Code: 1T46
Description: Imatinib also binds to the c-Kit receptor tyrosine kinase, which is overactive in certain cancers, including gastrointestinal stromal tumors (GIST). This structure shows imatinib bound to the ATP-binding site of c-Kit, inhibiting its kinase activity.
- PDGFR Kinase Bound to Imatinib
PDB Code: 1XBB
Description: Imatinib binds to the platelet-derived growth factor receptor (PDGFR) kinase, another kinase implicated in various cancers. This structure shows how imatinib inhibits PDGFR by binding to its inactive conformation.
- Abl Kinase Mutant (T315I) Bound to Imatinib
PDB Code: 3GVU
Description: This structure shows the Abl kinase with a T315I mutation, which is associated with resistance to imatinib in some cancer patients. This mutation alters the binding site, reducing imatinib’s effectiveness, and highlights mechanisms of drug resistance.
These structures illustrate how imatinib selectively stabilizes the inactive forms of kinases like Abl, c-Kit, and PDGFR, helping to control abnormal cell proliferation in various cancers.
Exploration 2: Do you know PDB codes for some example of an intrinsically disordered region bound to another protein?
This time I asked it "Do you know PDB codes for some example of an intrinsically disordered region bound to another protein?". And there it went:
ChatGPT said:
Yes, there are several well-characterized structures in the PDB where an intrinsically disordered region (IDR) of one protein binds to another protein. These IDRs typically adopt a defined structure upon binding, which is critical for their regulatory or signaling functions. Here are some well-known examples:
- p53 Transactivation Domain Bound to MDM2
PDB Code: 1YCR
Description: The N-terminal transactivation domain of p53, which is intrinsically disordered, binds to the MDM2 oncoprotein. This interaction regulates p53 stability and function.
- E1A from Adenovirus Bound to Rb
PDB Code: 1LXE
Description: The intrinsically disordered E1A protein from adenovirus binds to the retinoblastoma (Rb) protein, inhibiting Rb’s function in cell cycle control.
- C-Myc Transactivation Domain Bound to TAZ1 of CBP
PDB Code: 1SB0
Description: The disordered transactivation domain of c-Myc adopts an alpha-helical structure when it binds to the TAZ1 domain of CBP, a transcriptional co-activator.
- p27Kip1 Bound to Cyclin A/Cdk2
PDB Code: 1JSU
Description: The intrinsically disordered p27Kip1 binds and inhibits the Cyclin A/Cdk2 complex by wrapping around the cyclin and Cdk subunits. This interaction plays a key role in cell cycle regulation.
- Ash1 Intrinsically Disordered Region Bound to the Chromodomain of Polycomb
PDB Code: 2L3J
Description: The intrinsically disordered region of Ash1 interacts with the chromodomain of Polycomb (Pc), a regulator of transcriptional silencing, adopting a structured form upon binding.
- pKID of CREB Bound to KIX Domain of CBP
PDB Code: 1KDX
Description: The phosphorylated kinase-inducible domain (pKID) of CREB, which is intrinsically disordered, binds to the KIX domain of CBP. Phosphorylation induces an alpha-helix formation in pKID, stabilizing the interaction.
- Ets1 Transcription Factor Bound to DNA
PDB Code: 2NNY
Description: The disordered regulatory region of Ets1 becomes partially ordered upon binding DNA, an example of a disordered region that adopts structure when interacting with a non-protein partner.
- Hif-1α Oxygen-Dependent Degradation Domain Bound to VHL
PDB Code: 1LQB
Description: The oxygen-dependent degradation domain of Hif-1α, which is intrinsically disordered, binds to the von Hippel-Lindau (VHL) protein. This interaction marks Hif-1α for degradation under normoxic conditions.
- Tau Protein Bound to Microtubules
PDB Code: 5SYF
Description: Tau is an intrinsically disordered protein that interacts with microtubules. Upon binding, specific regions of Tau adopt structured conformations, helping to stabilize the microtubule assembly.
- Histone H1 Disordered Tail Bound to DNA or Histone Chaperones
PDB Code: 2LXD
Description: The disordered C-terminal tail of histone H1 binds DNA and histone chaperones, adopting partial structure upon binding. This interaction plays a role in chromatin compaction.
These structures showcase the "folding-upon-binding" property of many IDRs, where they adopt specific secondary or tertiary structures in the presence of their binding partners, often facilitating critical cellular processes like signaling, regulation, and gene expression.
Example: PDB 1YCR, including back-prediction with AlphaFold 3 using the full proteins
I then set myself to look at some of these examples hands-on. PDB 1YCR (first answer provided here by ChatGPT-4o) is a structure of p53, a protein related to cancer in various ways and that contains several disordered residues that bind to other proteins. Here in this PDB file, it was caught interacting with MDM2. You see this clear in the experimental structure at the top left of the figure, where a short segment from MDM2 binds onto p53 as a helix. And you also see this in the prediction from AlphaFold 3 for the full complex, which suggests some additional interactions.

Exploration 3: Propose PDB entries for proteins bound to others involving a post-translational modification?
ChatGPT said:
Certainly! Here are several PDB entries featuring proteins that are involved in interactions mediated by post-translational modifications (PTMs). These structures provide insights into how PTMs like phosphorylation, methylation, and acetylation influence protein-protein interactions.
- Phosphorylation Recognition
14–3–3 Protein Bound to Phosphorylated Peptide
PDB Code: 1QJA
Description: The structure shows the 14–3–3 protein bound to a phosphorylated peptide, illustrating how 14–3–3 recognizes and binds phosphoserine residues.
BRCT Domain Bound to Phosphorylated Peptide
PDB Code: 1T29
Description: This structure features the BRCA1 BRCT domain bound to a phosphorylated peptide from BACH1, demonstrating how phosphorylation is recognized by BRCT domains.
- Ubiquitin Recognition
Ubiquitin-Binding Domain of p62 Bound to Ubiquitin
PDB Code: 3B0F
Description: The structure depicts the UBA (ubiquitin-associated) domain of p62 bound to ubiquitin, highlighting how p62 recognizes and binds polyubiquitinated substrates for autophagy.
- Methylation Recognition
Chromodomain of HP1 Bound to Methylated Histone
PDB Code: 3C1A
Description: The HP1 chromodomain interacts with a trimethylated histone H3K9 peptide, showcasing how methylation on histones is recognized and contributes to transcriptional regulation.
- Acetylation Recognition
Acetylated Histone H4 Bound to Bromodomain
PDB Code: 3MXF
Description: This structure presents the bromodomain of the BET protein BRD4 bound to an acetylated histone H4 tail, illustrating how acetylation of histones is recognized and mediates chromatin interactions.
- Neddylation Recognition
cullin-RING E3 Ligase Bound to Nedd8
PDB Code: 2ZVJ
Description: The structure shows the interaction between a cullin-RING E3 ligase and the Nedd8 modification, highlighting the role of neddylation in the ubiquitin-proteasome system.
- Phosphorylation of p27Kip1 and Cyclin-Cdk Binding
Cdk2-cyclin A Complex Bound to Phosphorylated p27Kip1
PDB Code: 1JSU
Description: This structure shows the cyclin A/Cdk2 complex interacting with the phosphorylated form of p27Kip1, demonstrating how phosphorylation modulates the binding and inhibition of cyclin-dependent kinases.
- Glycosylation
Lectin Domain of Concanavalin A Bound to Mannosylated Glycoprotein
PDB Code: 3FQZ
Description: This structure features concanavalin A, a plant lectin, bound to a mannosylated glycoprotein, illustrating how glycosylation can mediate protein-protein interactions.
- Phosphorylation of Ets1 Transcription Factor
Ets1 Bound to DNA
PDB Code: 2NNY
Description: The disordered regulatory region of Ets1 is partially ordered upon binding DNA, showcasing how phosphorylation affects its ability to interact with target sequences.
- Acetylation of p53
Acetylated p53 Bound to MDM2
PDB Code: 1YCR
Description: The interaction between acetylated p53 and MDM2 illustrates how acetylation can affect the stability and regulation of p53, a critical tumor suppressor.
- SUMOylation
SUMO-1 Bound to a SUMO-Interacting Motif
PDB Code: 4C0N
Description: This structure shows a protein containing a SUMO-interacting motif bound to SUMO-1, highlighting how SUMOylation regulates various protein interactions.
Example: PDB 1QJA, including back-prediction with AlphaFold 3 (which can treat phosphorylated residues!)
PDB 1QJA (first answer provided by ChatGPT-4o) is a structure of the protein called 14–3–3 bound to a peptide with a phosphorylation (P atom surrounded by 4 O atoms) in it. In the picture below, the 3D structures correspond to the complex observed between the protein and the phosphorylated peptide (left: full view, right: zoom on the phosphate). You can see how the phosphate group establishes several contacts (dashed lines). The green 2D plots below are AlphaFold 3’s PAE plots, showing better statistics (more green) for the peptide interacting with the protein when the phosphate group is present (right).

Did GPT-4o Really Learn the PDB Upon Training?
Since OpenAI didn’t answer to this when I asked by email, I just asked ChatGPT-4o itself. And the answer is… kind of, but not quite!
What I got from ChatGPT is that it was not explicitly trained with PDB entries (which would have been nice, because they contain paper abstracts and tons of other data for each PDB entry). However, ChatGPT explained that its training material included papers, preprints, abstracts, and educational documents that referred to these PDB entries; and that that’s how it got to know about a huge number of PDB entries.
Hallucinations of PDB entries, and a way to overcome them
If the above is correct, that is if GPT-4o wasn’t trained with the PDB itself but with papers and other documents referring to certain PDB structures, then it might perfectly happen that it doesn’t know about many structures, especially those that aren’t linked to any paper. Following this idea, I set myself to interrogate ChatGPT-4o a bit deeper and trying to expose it.
It took me some time to crack it, but eventually I managed. On my first question like "Do you know PDB ID 1CI0?" (which the PDB shows not linked to any paper), I got a response confidently but wrongly explaining that this entry "describes a cytochrome c553 from Desulfovibrio vulgaris". The PDB entry however explains that this is another protein that is neither a cytochrome c nor does it belong to Desulfovibrio vulgaris.
Now that’s bad, very bad like with all hallucinations. But encouragingly, if I ask ChatGPT-4o to do an online search (note that the PDB is totally open), then it distils the correct information:
The PDB ID 1CI0 refers to the crystal structure of pyridoxine-5′-phosphate (PNP) oxidase from Saccharomyces cerevisiae (brewer’s yeast). This enzyme is classified as an oxidoreductase and plays a key role in the vitamin B6 metabolic pathway. It facilitates the conversion of pyridoxine-5′-phosphate to pyridoxal-5′-phosphate, an active form of vitamin B6.
The structure was determined using X-ray diffraction at a resolution of 2.7 Å, with the biological assembly being a homo-dimer. The experiment was conducted using crystals derived from a recombinant protein expressed in Escherichia coli. The structure includes two protein chains and is bound to the cofactor flavin mononucleotide (FMN), which is crucial for its enzymatic activity.
This entry was deposited in 1999 by Shi et al. and is often used to study vitamin B6 biosynthesis mechanisms and potential applications in biotechnology or medicine【7】【8】【10】.
…which essentially reflects perfectly all the information available for this entry at the PDB and it’s actually nicer than it because it spells out acronym and short-form names like "PNP" and cleans up all the data – just compare with the PDB entry:

So? Useful or not?
My conclusion with this latest experiment is that despite being perfectly prone to hallucination, cross-checking by forcing ChatGPT to back-search the entries online could probably totally overcome its hallucination problems.
Like with all these AI tools, we need caution, but the search-back approach is probably reliable— yet before drawing very specific conclusions, you better cross-check the LLM’s output yourself.
Conclusion and Prospects
Through this exploration, I aimed to illustrate how experimental structural biology, molecular modeling with tools like AlphaFold 3, and LLMs like GPT-4o, can converge, enabling researchers to search and analyze molecular structures in novel ways, all thanks to OpenAI including content about the PDB in its training dataset. From the hallucination problems we saw in the last section, I propose that including information for PDB entries explicitly upon LLM training could take all this to a new level, working better and more accurately. Yet with the search-back approach tested above, one can probably work feeling safe that the LLM won’t be sneak in false information.
I think that by leveraging these combined resources, scientists can much faster and better get acquainted with the range of structures available in connection to a given topic; probably most useful when moving into a new specific subdomain of biology.
I also think that these resources lay the groundwork for a more through investigation of how LLMs and AlphaFold 3 (or similar models that are emerging now) could be coupled to not just navigate but also understand biomolecules and their complexes in new ways. Perhaps even molecular graphics and modeling tools that benefit from an LLMs’ knowledge of the PDB could also be created that allow to perform complex manipulation and analyses of biomolecular structures through natural commands.
www.lucianoabriata.com I write about everything that lies in my broad sphere of interests: nature, science, technology, programming, etc. Subscribe to get my new stories by email. To consult about small jobs check my services page here. You can contact me here. You can tip me here.
The above is the detailed content of Epic “Crossover” Between AlphaFold 3 and GPT-4o’s Knowledge of Protein Data Bank Entries. For more information, please follow other related articles on the PHP Chinese website!

 10 Applications of LLM Agents for BusinessApr 13, 2025 am 09:34 AM
10 Applications of LLM Agents for BusinessApr 13, 2025 am 09:34 AMIntroduction Large language models or LLMs are a game-changer especially when it comes to working with content. From supporting summarization, translation, and generation, LLMs like GPT-4, Gemini, and Llama have made it simple
 How LLM Agents are Reshaping Workplace?Apr 13, 2025 am 09:33 AM
How LLM Agents are Reshaping Workplace?Apr 13, 2025 am 09:33 AMIntroduction Large language model (LLM) agents are the latest innovation boosting workplace business efficiency. They automate repetitive activities, boost collaboration, and provide useful insights across departments. Unlike
 Setup Mage AI with PostgresApr 13, 2025 am 09:31 AM
Setup Mage AI with PostgresApr 13, 2025 am 09:31 AMImagine yourself as a data professional tasked with creating an efficient data pipeline to streamline processes and generate real-time information. Sounds challenging, right? That’s where Mage AI comes in to ensure that the lende
 Is Google's Imagen 3 the Future of AI Image Creation?Apr 13, 2025 am 09:29 AM
Is Google's Imagen 3 the Future of AI Image Creation?Apr 13, 2025 am 09:29 AMIntroduction Text-to-image synthesis and image-text contrastive learning are two of the most innovative multimodal learning applications recently gaining popularity. With their innovative applications for creative image creati
 Top 10 YouTube Channels to Learn Excel - Analytics VidhyaApr 13, 2025 am 09:27 AM
Top 10 YouTube Channels to Learn Excel - Analytics VidhyaApr 13, 2025 am 09:27 AMIntroduction Excel is indispensable for boosting productivity and efficiency across all the fields. The wide range of resources on YouTube can help learners of all levels find helpful tutorials specific to their needs. This ar
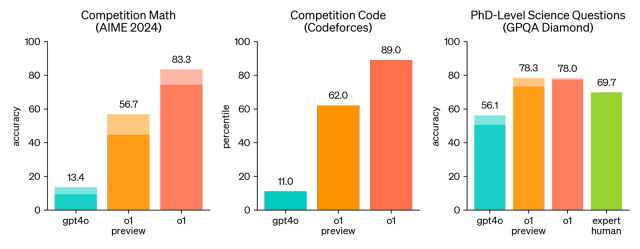 OpenAI o1: A New Model That 'Thinks' Before Answering ProblemsApr 13, 2025 am 09:26 AM
OpenAI o1: A New Model That 'Thinks' Before Answering ProblemsApr 13, 2025 am 09:26 AMHave you heard the big news? OpenAI just rolled out preview of a new series of AI models – OpenAI o1 (also known as Project Strawberry/Q*). These models are special because they spend more time “thinking” befor
 Claude vs Gemini: The Comprehensive Comparison - Analytics VidhyaApr 13, 2025 am 09:20 AM
Claude vs Gemini: The Comprehensive Comparison - Analytics VidhyaApr 13, 2025 am 09:20 AMIntroduction Within the quickly changing field of artificial intelligence, two language models, Claude and Gemini, have become prominent competitors, each providing distinct advantages and skills. Although both models can mana
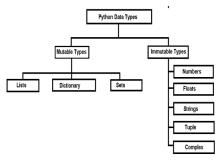 Mutable vs Immutable Objects in Python - Analytics VidhyaApr 13, 2025 am 09:15 AM
Mutable vs Immutable Objects in Python - Analytics VidhyaApr 13, 2025 am 09:15 AMIntroduction Python is an object-oriented programming language (or OOPs).In my previous article, we explored its versatile nature. Due to this, Python offers a wide variety of data types, which can be broadly classified into m


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

SecLists
SecLists is the ultimate security tester's companion. It is a collection of various types of lists that are frequently used during security assessments, all in one place. SecLists helps make security testing more efficient and productive by conveniently providing all the lists a security tester might need. List types include usernames, passwords, URLs, fuzzing payloads, sensitive data patterns, web shells, and more. The tester can simply pull this repository onto a new test machine and he will have access to every type of list he needs.

PhpStorm Mac version
The latest (2018.2.1) professional PHP integrated development tool

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

DVWA
Damn Vulnerable Web App (DVWA) is a PHP/MySQL web application that is very vulnerable. Its main goals are to be an aid for security professionals to test their skills and tools in a legal environment, to help web developers better understand the process of securing web applications, and to help teachers/students teach/learn in a classroom environment Web application security. The goal of DVWA is to practice some of the most common web vulnerabilities through a simple and straightforward interface, with varying degrees of difficulty. Please note that this software

SublimeText3 Mac version
God-level code editing software (SublimeText3)

