
 Technology peripheralsAIDefeating 25 molecular design algorithms, Georgia Tech, University of Toronto, and Cornell proposed large language model MOLLEO
Technology peripheralsAIDefeating 25 molecular design algorithms, Georgia Tech, University of Toronto, and Cornell proposed large language model MOLLEO
Author | Wang Haorui, Georgia Institute of Technology
Editor | ScienceAI
Molecular discovery as an optimization problem poses significant computational challenges because the optimization objective may not be differentiable. Evolutionary algorithms (EAs) are commonly used to optimize black-box targets in molecular discovery by traversing chemical space through random mutation and crossover, but this results in extensive and expensive target evaluation.
In this work, researchers from the Georgia Institute of Technology, the University of Toronto, and Cornell University collaborated to propose Molecular Language Enhanced Evolutionary Optimization (MOLLEO), which integrates pre-trained large language models (LLMs) with chemical knowledge into evolutionary optimization. In the algorithm, the molecular optimization capability of the evolutionary algorithm has been significantly improved.
The study, titled "Efficient Evolutionary Search Over Chemical Space with Large Language Models", was published on the preprint platform arXix on June 23.
Paper link: https://arxiv.org/abs/2406.16976
The huge computational challenge of molecular discovery
Molecular discovery is a complex iterative process involving the design, synthesis, and Evaluation and improvement have a wide range of real-world applications, including drug design, materials design, improving energy, disease problems, etc. This process is often slow and laborious, and even approximate computational evaluations require significant resources due to complex design conditions and evaluation of molecular properties that often require expensive evaluations (such as wet experiments, bioassays, and computational simulations).
Therefore, developing efficient molecular search, prediction and generation algorithms has become a research hotspot in the field of chemistry to accelerate the discovery process. In particular, machine learning-driven methods have played an important role in rapidly identifying and proposing promising molecular candidates.
Due to the importance of the problem, molecular optimization has received great attention, including more than 20 molecular design algorithms that have been developed and tested (among them, combinatorial optimization methods such as genetic algorithms and reinforcement learning are ahead of other generative models and continuous optimization algorithms ), Please refer to the recent review article of the Naturesub-journal for details. One of the most effective methods is evolutionary algorithms (EAs). The characteristic of these algorithms is that they do not require gradient evaluation, so they are very suitable for black-box objective optimization in molecular discovery.
However, a major drawback of these algorithms is that they randomly generate candidate structures without exploiting task-specific information, resulting in the need for extensive objective function evaluation. Because evaluating attributes is expensive, molecular optimization not only finds the molecular structure with the best expected attributes, but also minimizes the number of evaluations of the objective function (which is also equivalent to improving search efficiency).
Recently, LLM has demonstrated some basic capabilities in multiple chemistry-related tasks, such as predicting molecular properties, retrieving optimal molecules, automating chemical experiments, and generating molecules with target properties. Since LLMs are trained on large-scale text corpora covering a wide range of tasks, they demonstrate general language understanding capabilities and basic chemical knowledge, making them an interesting tool for chemical discovery tasks.
However, many LLM-based methods rely on in-context learning and cue engineering, which can be problematic when designing molecules with strict numerical goals, as LLM may have difficulty meeting precise numerical constraints or optimizing specific numerical target. Furthermore, methods that rely solely on LLM hints may generate molecules with poor physical foundation or generate invalid SMILES strings that cannot be decoded into chemical structures.
Molecular Language Enhanced Evolutionary Optimization
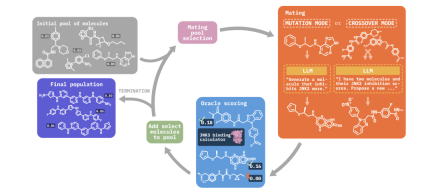
In this study, we propose Molecular Language Enhanced Evolutionary Optimization (MOLLEO), which integrates LLM into EA to improve the quality of generated candidates and accelerate the optimization process. MOLLEO utilizes LLM as a genetic operator to generate new candidates through crossover or mutation. We demonstrate for the first time how LLM can be integrated into the EA framework for molecule generation.
In this study, we considered three language models with different capabilities: GPT-4, BioT5, and MoleculeSTM. We integrate each LLM into different crossover and mutation procedures and demonstrate our design choices through ablation studies.
We have proven the superior performance of MOLLEO through experiments on multiple black-box optimization tasks, including single-objective and multi-objective optimization. For all tasks, including the more challenging protein-ligand docking, MOLLEO outperforms baseline EA and 25 other strong baseline methods. Additionally, we demonstrate the ability of MOLLEO to further optimize on the best JNK3 inhibitor molecules in the ZINC 250K database.
Our MOLLEO framework is based on a simple evolutionary algorithm, the Graph-GA algorithm, and enhances its functionality by integrating chemically aware LLM in genetic operations.
We first outline the problem statement, emphasizing the need to minimize expensive objective evaluations in black-box optimization. MOLLEO utilizes LLMs such as GPT-4, BioT5, and MoleculeSTM to generate new candidate molecules guided by target descriptions.
Specifically, in the crossover step, instead of randomly combining two parent molecules, we use LLM to generate molecules that maximize the target fitness function. In the mutation step, the operator mutates the fittest member of the current population according to the target description. However, we noticed that LLM did not always generate candidates with higher fitness than the input molecules, so we constructed selection pressures to filter edited molecules based on structural similarity.
Experimental results
We evaluated MOLLEO on 18 tasks. Tasks are selected from PMO and TDC benchmarks and databases and can be divided into the following categories:
- Structure-based optimization: Optimize molecules according to the target structure, including isomers generation based on the target molecule formula (isomers_c9h10n2o2pf2cl) and Two tasks based on matching or avoiding scaffold and substructural motifs (deco_hop, scaffold_hop).
- Name-based optimization: Includes finding compounds similar to known drugs (mestranol_similarity, thiothixene_rediscovery) and three multi-attribute optimization tasks (MPO) that rediscover drugs while rediscovering them (e.g. Perindopril, Ranolazine, Sitagliptin) Optimize other properties such as hydrophobicity (LogP) and permeability (TPSA). Although these tasks primarily involved the rediscovery of existing drugs rather than the design of new molecules, they demonstrated LLM's fundamental chemical optimization capabilities.
- Property Optimization: Includes the simple property optimization task QED, which measures the drug similarity of molecules. We then focused on three tasks in the PMO, measuring the activity of molecules against the following proteins: DRD2 (dopamine receptor D2), GSK3β (glycogen synthase kinase-3β), and JNK3 (c-Jun N-terminal kinase-3) . Additionally, we include three protein-ligand docking tasks in TDC (structural drug design) that are closer to real-world drug design than simple physicochemical properties.
To evaluate our method, we follow the PMO benchmark method, taking into account the target value and computing budget, and report the area under the curve (AUC top-k) of the top k average attribute values and the number of target function calls.
As a comparison benchmark, we used the top models in the PMO benchmark, including REINVENT based on reinforcement learning, the basic evolutionary algorithm Graph-GA and the Gaussian process Bayesian optimization GP BO.

Illustration: Top-10 AUC of single-target tasks. (Source: paper)
We conducted single-objective optimization experiments on 12 tasks of PMO. The results are shown in the table above. We report the AUC top-10 score of each task and the overall ranking of each model. The results show that using any large language model (LLM) as a genetic operator can improve performance beyond the default Graph-GA and all other baseline models.
GPT-4 outperformed all models in 9 out of 12 tasks, demonstrating its effectiveness and prospects as a general large language model in molecule generation. BioT5 achieved the second best result among all test models, with a total score close to GPT-4, indicating that small models trained and fine-tuned on domain knowledge also have good application prospects in MOLLEO.
MOLSTM is a small model based on the CLIP model that is fine-tuned on the natural language description of the molecule and the chemical formula of the molecule. We use the gradient descent algorithm in the evolutionary algorithm on the same natural language description to generate different new molecules, and its performance is also outperforms other baseline methods.

Illustration: JNK3 inhibits the population fitness that occurs as the number of iterations increases. (Source: paper)
To verify the effectiveness of integrating LLM into the EA framework, we show the score distribution of the initial random molecule pool on the JNK3 task. Subsequently, we performed a round of editing on all molecules in the pool and plotted the JNK3 score distribution of the edited molecules.
The results show that the distributions edited by LLM are all slightly shifted towards higher scores, indicating that LLM does provide useful modifications. However, the overall target score is still low, so single-step editing is not sufficient and iterative optimization using evolutionary algorithms is necessary here.

Illustration: The average docking score of the top 10 molecules when docked with DRD3, EGFR or adenosine A2A receptor protein. (Source: paper)
In addition to the 12 single-objective optimization tasks in PMO, we also tested MOLLEO on more challenging protein-ligand docking tasks, which are closer to real-world molecule generation scenarios than single-objective tasks. The above figure is a plot of the average docking score of the top ten best molecules of MOLLEO and Graph-GA versus the number of target function calls.
The results show that in all three proteins, the docking scores of molecules generated by our method are almost always better than those of the baseline model and the convergence speed is faster. Among the three language models we used, BioT5 performed best. In reality, better docking scores and faster convergence rates can reduce the number of bioassays required to screen molecules, making the process more cost- and time-effective.

Illustration: Sum and hypervolume fraction for multi-objective tasks. (Source: paper)

Illustration: Pareto optimal visualization of Graph-GA and MOLLEO on multi-objective tasks. (Source: paper)
For multi-objective optimization, we consider two metrics: AUC top-10 of the sum of scores of all optimization objectives and the hypervolume of the Pareto optimal set. We present the results of multi-objective optimization on three tasks. Tasks 1 and 2 are inspired by drug discovery goals and aim to optimize three goals simultaneously: maximizing a molecule's QED, minimizing its synthetic accessibility (SA) score (meaning easier to synthesize), and maximizing its contribution to JNK3 (Task 1) or GSK3β (Task 2) binding scores. Task 3 is more challenging because it requires simultaneous optimization of five objectives: maximizing QED and JNK3 binding scores, and minimizing GSK3β binding scores, DRD2 binding scores, and SA scores.
We find that MOLLEO (GPT-4) consistently outperforms the baseline Graph-GA in both hypervolume and summation across all three tasks. In the figure, we visualize the Pareto optimal sets (in the objective space) of our method and Graph-GA in Task 1 and Task 2. The performance of open source language models decreases when multiple targets are introduced. We speculate that this performance degradation may stem from their inability to capture large amounts of information-dense context.

Illustration: Initializing MOLLEO using the best molecules in ZINC 250K. (Source: paper)
The ultimate goal of the evolutionary algorithm is to improve the properties of the initial molecule pool and discover new molecules. In order to explore the ability of MOLLEO to explore new molecules, we initialize the molecule pool with the best molecules in ZINC 250K, and then use MOLLEO and Graph-GA for optimization. Experimental results on the JNK3 task show that our algorithm consistently outperforms the baseline model Graph-GA and is able to improve on the best molecules found in existing datasets.
In addition, we also noticed that the training set of BioT5 is the ZINC20 database (containing 1.4 billion compounds), and the training set of MoleculeSTM is the PubChem database (about 250,000 molecules). We checked whether the final molecules generated by each model in the JNK3 task appeared in the corresponding dataset. It was found that the generated molecules did not overlap with those in the data set. This shows that the model is able to generate new molecules that were not present in the training set.
Can be applied to drug discovery, materials, biomolecule design
Molecular discovery and design is a rich field with numerous practical applications, many beyond the scope of the current study but still relevant to our proposed framework. MOLLEO combines LLM with EA algorithms to provide a flexible algorithm framework through pure text. In the future, MOLLEO can be applied to scenarios such as drug discovery, expensive computer simulations, and the design of materials or large biomolecules.
Future work We will further focus on how to improve the quality of generated molecules, including their target values and discovery speed. As LLM continues to advance, we expect that the performance of the MOLLEO framework will also continue to improve, making it a promising tool in generative chemistry applications.
The above is the detailed content of Defeating 25 molecular design algorithms, Georgia Tech, University of Toronto, and Cornell proposed large language model MOLLEO. For more information, please follow other related articles on the PHP Chinese website!

 五个时间序列预测的深度学习模型对比总结May 05, 2023 pm 05:16 PM
五个时间序列预测的深度学习模型对比总结May 05, 2023 pm 05:16 PMMakridakisM-Competitions系列(分别称为M4和M5)分别在2018年和2020年举办(M6也在今年举办了)。对于那些不了解的人来说,m系列得比赛可以被认为是时间序列生态系统的一种现有状态的总结,为当前得预测的理论和实践提供了经验和客观的证据。2018年M4的结果表明,纯粹的“ML”方法在很大程度上胜过传统的统计方法,这在当时是出乎意料的。在两年后的M5[1]中,最的高分是仅具有“ML”方法。并且所有前50名基本上都是基于ML的(大部分是树型模型)。这场比赛看到了LightG
 RLHF与AlphaGo核心技术强强联合,UW/Meta让文本生成能力再上新台阶Oct 27, 2023 pm 03:13 PM
RLHF与AlphaGo核心技术强强联合,UW/Meta让文本生成能力再上新台阶Oct 27, 2023 pm 03:13 PM在一项最新的研究中,来自UW和Meta的研究者提出了一种新的解码算法,将AlphaGo采用的蒙特卡洛树搜索算法(Monte-CarloTreeSearch,MCTS)应用到经过近端策略优化(ProximalPolicyOptimization,PPO)训练的RLHF语言模型上,大幅提高了模型生成文本的质量。PPO-MCTS算法通过探索与评估若干条候选序列,搜索到更优的解码策略。通过PPO-MCTS生成的文本能更好满足任务要求。论文链接:https://arxiv.org/pdf/2309.150
 MIT团队运用机器学习闭环自主分子发现平台,成功发现、合成和描述了303种新分子Jan 04, 2024 pm 05:38 PM
MIT团队运用机器学习闭环自主分子发现平台,成功发现、合成和描述了303种新分子Jan 04, 2024 pm 05:38 PM编辑|X传统意义上,发现所需特性的分子过程一直是由手动实验、化学家的直觉以及对机制和第一原理的理解推动的。随着化学家越来越多地使用自动化设备和预测合成算法,自主研究设备越来越接近实现。近日,来自MIT的研究人员开发了由集成机器学习工具驱动的闭环自主分子发现平台,以加速具有所需特性的分子的设计。无需手动实验即可探索化学空间并利用已知的化学结构。在两个案例研究中,该平台尝试了3000多个反应,其中1000多个产生了预测的反应产物,提出、合成并表征了303种未报道的染料样分子。该研究以《Autonom
 AI助力脑机接口研究,纽约大学突破性神经语音解码技术,登Nature子刊Apr 17, 2024 am 08:40 AM
AI助力脑机接口研究,纽约大学突破性神经语音解码技术,登Nature子刊Apr 17, 2024 am 08:40 AM作者|陈旭鹏编辑|ScienceAI由于神经系统的缺陷导致的失语会导致严重的生活障碍,它可能会限制人们的职业和社交生活。近年来,深度学习和脑机接口(BCI)技术的飞速发展为开发能够帮助失语者沟通的神经语音假肢提供了可行性。然而,神经信号的语音解码面临挑战。近日,约旦大学VideoLab和FlinkerLab的研究者开发了一个新型的可微分语音合成器,可以利用一个轻型的卷积神经网络将语音编码为一系列可解释的语音参数(例如音高、响度、共振峰频率等),并通过可微分神经网络将这些参数合成为语音。这个合成器
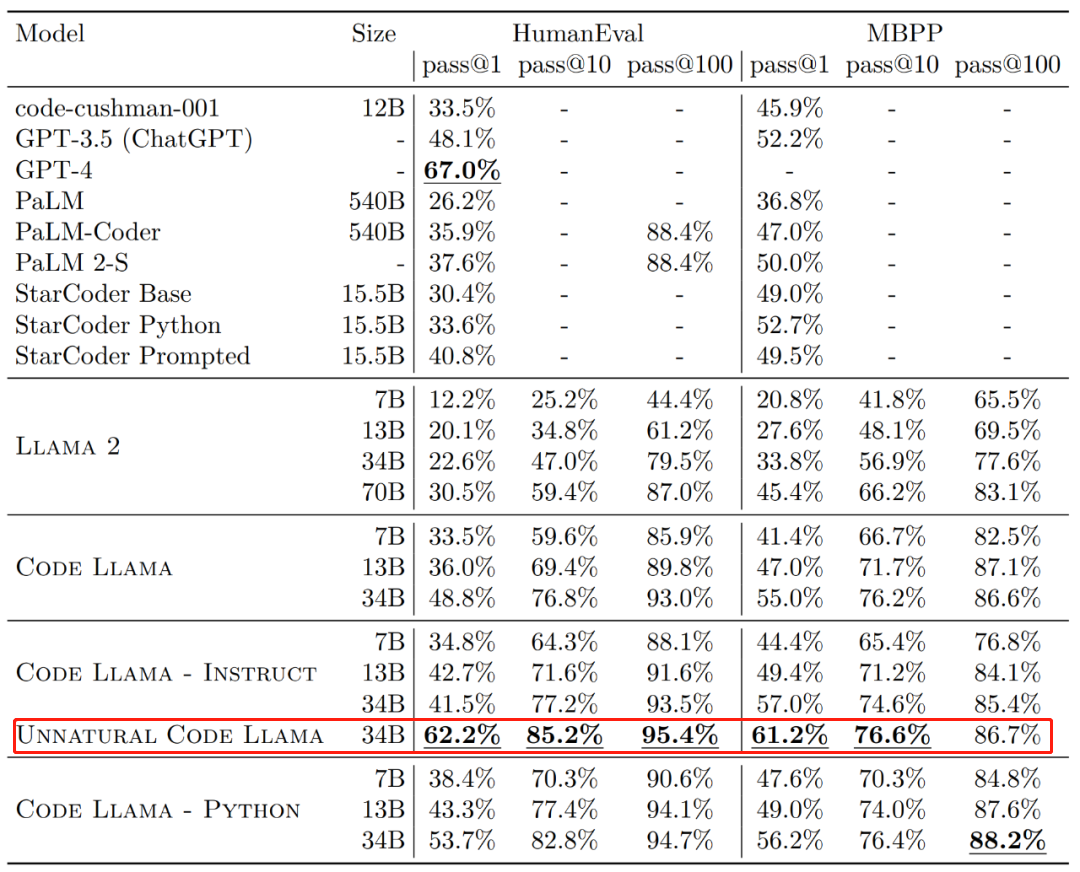 Code Llama代码能力飙升,微调版HumanEval得分超越GPT-4,一天发布Aug 26, 2023 pm 09:01 PM
Code Llama代码能力飙升,微调版HumanEval得分超越GPT-4,一天发布Aug 26, 2023 pm 09:01 PM昨天,Meta开源专攻代码生成的基础模型CodeLlama,可免费用于研究以及商用目的。CodeLlama系列模型有三个参数版本,参数量分别为7B、13B和34B。并且支持多种编程语言,包括Python、C++、Java、PHP、Typescript(Javascript)、C#和Bash。Meta提供的CodeLlama版本包括:代码Llama,基础代码模型;代码羊-Python,Python微调版本;代码Llama-Instruct,自然语言指令微调版就其效果来说,CodeLlama的不同版
 准确率 >98%,基于电子密度的 GPT 用于化学研究,登 Nature 子刊Mar 27, 2024 pm 02:16 PM
准确率 >98%,基于电子密度的 GPT 用于化学研究,登 Nature 子刊Mar 27, 2024 pm 02:16 PM编辑|紫罗可合成分子的化学空间是非常广阔的。有效地探索这个领域需要依赖计算筛选技术,比如深度学习,以便快速地发现各种有趣的化合物。将分子结构转换为数字表示形式,并开发相应算法生成新的分子结构是进行化学发现的关键。最近,英国格拉斯哥大学的研究团队提出了一种基于电子密度训练的机器学习模型,用于生成主客体binders。这种模型能够以简化分子线性输入规范(SMILES)格式读取数据,准确率高达98%,从而实现对分子在二维空间的全面描述。通过变分自编码器生成主客体系统的电子密度和静电势的三维表示,然后通
 手机摄影技术让以假乱真的好莱坞级电影特效视频走红Sep 07, 2023 am 09:41 AM
手机摄影技术让以假乱真的好莱坞级电影特效视频走红Sep 07, 2023 am 09:41 AM一个普通人用一台手机就能制作电影特效的时代已经来了。最近,一个名叫Simulon的3D技术公司发布了一系列特效视频,视频中的3D机器人与环境无缝融合,而且光影效果非常自然。呈现这些效果的APP也叫Simulon,它能让使用者通过手机摄像头的实时拍摄,直接渲染出CGI(计算机生成图像)特效,就跟打开美颜相机拍摄一样。在具体操作中,你要先上传一个3D模型(比如图中的机器人)。Simulon会将这个模型放置到你拍摄的现实世界中,并使用准确的照明、阴影和反射效果来渲染它们。整个过程不需要相机解算、HDR
 谷歌用大型模型训练机器狗理解模糊指令,激动不已准备去野餐Jan 16, 2024 am 11:24 AM
谷歌用大型模型训练机器狗理解模糊指令,激动不已准备去野餐Jan 16, 2024 am 11:24 AM人类和四足机器人之间简单有效的交互是创造能干的智能助理机器人的途径,其昭示着这样一个未来:技术以超乎我们想象的方式改善我们的生活。对于这样的人类-机器人交互系统,关键是让四足机器人有能力响应自然语言指令。近来大型语言模型(LLM)发展迅速,已经展现出了执行高层规划的潜力。然而,对LLM来说,理解低层指令依然很难,比如关节角度目标或电机扭矩,尤其是对于本身就不稳定、必需高频控制信号的足式机器人。因此,大多数现有工作都会假设已为LLM提供了决定机器人行为的高层API,而这就从根本上限制了系统的表现能


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

AI Hentai Generator
Generate AI Hentai for free.

Hot Article
Hot Tools

ZendStudio 13.5.1 Mac
Powerful PHP integrated development environment

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

EditPlus Chinese cracked version
Small size, syntax highlighting, does not support code prompt function

DVWA
Damn Vulnerable Web App (DVWA) is a PHP/MySQL web application that is very vulnerable. Its main goals are to be an aid for security professionals to test their skills and tools in a legal environment, to help web developers better understand the process of securing web applications, and to help teachers/students teach/learn in a classroom environment Web application security. The goal of DVWA is to practice some of the most common web vulnerabilities through a simple and straightforward interface, with varying degrees of difficulty. Please note that this software

Atom editor mac version download
The most popular open source editor

