
 Technology peripheralsAIThe success rate exceeds that of the RoseTTAFold series, using sequence information to directly predict protein-ligand complex structures.
Technology peripheralsAIThe success rate exceeds that of the RoseTTAFold series, using sequence information to directly predict protein-ligand complex structures.

Editor | Radish Skin
Protein is a well-established tool in the body's fight against pathogens and is used to narrow down the range of potential treatments for experimental testing. High-quality protein structure is required, and proteins are often viewed as fully or partially rigid.
Here, researchers at Freie Universität Berlin have developed an artificial intelligence system that can predict fully flexible all-atom structures of protein-ligand complexes directly from sequence information.
Although classical docking methods are still superior, this also depends on the crystal structure of the target protein. In addition to predicting flexible all-atom structures, the prediction confidence metric (plDDT) can be used to select accurate predictions and differentiate between strong and weak binders.
The study is titled "Structure prediction of protein-ligand complexes from sequence information with Umol" and was published in "Nature Communications on May 28, 2024 》.

#Protein-protein target contacts are important issues in evaluating new drugs and repurposing known ones. Existing contact methods have limitations: they require high-quality protein structures; it is difficult to determine accurate contact postures; they are mostly based on binding ability (affinity) evaluation, which is difficult to reflect other factors such as structural stability. However, existing contact methods are limited by the need for high-quality protein structures, accurate contact poses, and multi-based affinity assessment. Therefore, the exploration of new ligands is limited by a combined approach of protein assembly and structure evaluation.
Although machine learning has been applied in this field, its performance on known target areas has not surpassed the classic methods based on scoring functions. Moreover, the predicted protein structure is often not suitable for direct use in ligand docking.
In addition, if the structures in the evaluation set are divided based on release time rather than similarity, bias will be introduced, especially when facing receptor structures not seen in training, the performance will be halved.
Protein flexibility is crucial for reaching the binding state and successful docking. Although RoseTTAFold All-Atom can bind ligands when predicting proteins, its success rate on the PoseBusters test set is only 42%, and it is The behavior of unseen proteins is unknown, indicating that the challenge of protein-ligand complex structure prediction has not yet been fully resolved.
A team at Freie Universität Berlin has developed an AI method that can predict the structure of protein-ligand complexes based on sequence information by extending EvoFormer in AlphaFold2. This network is similar to RFAA except that 3D trajectories are not included and template structures or additional crystallographic ligand data are used as input or during training.
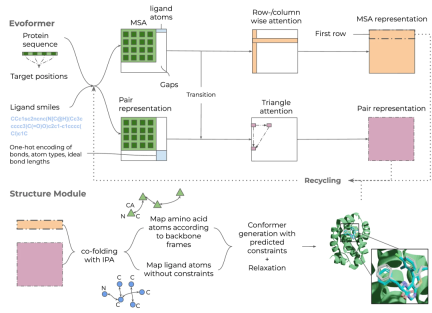
Illustration: Umol overview. (Source: Paper)
Starting from a protein sequence, alternative protein target sites (pockets), and ligands SMILES creates a multiple sequence alignment (MSA) and bond matrix. From this, features are generated within the network and 3D structures are generated. Since no structural information is required to generate the final protein-ligand complex structure, there are no restrictions on protein or ligand flexibility.
Umol achieves a higher success rate (SR, ligand RMSD ≤ 2 Å) when including pocket information on the PoseBusters test set compared to the closest RoseTTAFold All-Atom and NeuralPlexer1, 45 respectively %, 42%, and 24%, making it the best performing method in protein-ligand structure prediction.

Illustration: Prediction accuracy. (Source: paper)
When pocket information is removed from Umol and template information is removed from RFAA, the SR drops to 18% and 8% respectively. When using DiffDock with AF prediction, the accuracy is 21% but depends on highly accurate interface prediction (pocket RMSD
Many ligand poses slightly above the 2 Å success threshold may be comparable, suggesting that a more flexible scoring system may be needed. Umol's success rate exceeds AutoDock Vina at the 2.35 Å threshold. Even small alignment errors can become problematic when native protein structures are not used for scoring.
Cofolded protein-ligand complexes have the potential to accelerate drug repositioning. In particular, the researchers found that the predicted lDDT of the ligand (plDDT) can be used to select accurate docking poses, while the pIDDT of the protein pocket is suitable for selecting accurate interfaces.

Illustration: Confidence metrics and accuracy. (Source: Paper)
ligand plDDT also separated high-affinity ligands from low-affinity ligands, suggesting that some of the predictions for Umol and Umol-pocket uncertainty may be weak binders. This further demonstrates the capabilities of Umol and highlights that important aspects of protein-ligand interactions appear to be understood.

Illustration: BindingDB prediction. (Source: paper)
Despite the 18% accuracy without pocket information, the network can still differentiate between strong and weak binders to a certain extent. This is particularly useful for annotating unknown complexes, and the team presented 336 protein-ligand structures with very high confidence (ligand plDDT>85). It should be noted that although these structures appear reasonable and their L-plDDT scores are high, they still need to be verified experimentally.
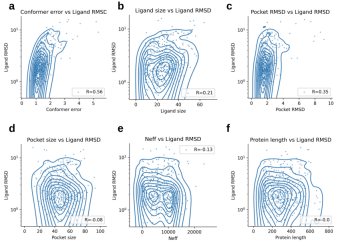
Illustration: Using Umol-pocket to analyze the relationship between predicted different features and ligand RMSD (LRMSD) on the PoseBusters test set (n=428). (Source: Paper)
The researchers did not find a clear relationship between the model's predictive performance and "different features associated with the same protein or ligand."

Illustration: The 5 most difficult structures. (Source: paper)
However, Umol-pocket was accurate in 3 out of 5 cases where other methods were difficult to predict. By inverting the trained network, new ligand-binding proteins or protein-binding ligands can be designed. Another option is to use transfer learning to create a generative diffusion model for the same purpose. In this case, the ligand or protein plDDT can be maximized in an attempt to create a high-affinity binder.
The current version of PDBbind contains data processed from the PDB in 2019. Since then, additional protein-ligand complexes have been submitted, suggesting that greater accuracy may be achievable.
However, it is currently unclear what precision is required to obtain meaningful protein-ligand docking results. The high accuracy of protein structure prediction is not achievable in tasks involving other molecules, such as small molecules or RNA.
Without protein co-evolutionary information, the accuracy of structure predictions rapidly decreases. Since there are no similar sources of information for small molecules or RNA, one has to rely on atomic representations.
Table: Success rate (percentage of ligands with RMSD≤2Å) on the PoseBuster benchmark set divided by sequence identity (seqid) for the PDBBind 2020 version. (Source: Paper)

# Researchers believe that pocket information is very effective, and without pocket information, deep learning methods seem prone to overfitting. This finding further corroborates the observation that although many molecules in the PoseBusters test set contain highly similar analogues in the training data set, this similarity does not correlate with model success.

Illustration: Some tests. (Source: Paper)
The same degree of overfitting is not observed for structure-based docking methods such as Vina or Gold. This is expected since they are based on atomic scoring functions and therefore do not rely on protein homology to the same extent.
The deep learning method has significantly higher performance on the training set, indicating that protein homology plays an important role in protein-ligand docking. The performance of RFAA on the test set is higher than that on the training set, which indicates possible data leakage between the training and test sets.
In summary, we are still a long way from fully grasping the complexity of protein-ligand interactions, but using deep learning to predict the structure of the entire complex may bring scientists closer to a solution.
Umol: https://github.com/patrickbryant1/Umol
Paper link: https://www.nature.com/articles/s41467 -024-48837-6
The above is the detailed content of The success rate exceeds that of the RoseTTAFold series, using sequence information to directly predict protein-ligand complex structures.. For more information, please follow other related articles on the PHP Chinese website!

 As AI Use Soars, Companies Shift From SEO To GEOMay 05, 2025 am 11:09 AM
As AI Use Soars, Companies Shift From SEO To GEOMay 05, 2025 am 11:09 AMWith the explosion of AI applications, enterprises are shifting from traditional search engine optimization (SEO) to generative engine optimization (GEO). Google is leading the shift. Its "AI Overview" feature has served over a billion users, providing full answers before users click on the link. [^2] Other participants are also rapidly rising. ChatGPT, Microsoft Copilot and Perplexity are creating a new “answer engine” category that completely bypasses traditional search results. If your business doesn't show up in these AI-generated answers, potential customers may never find you—even if you rank high in traditional search results. From SEO to GEO – What exactly does this mean? For decades
 Big Bets On Which Of These Pathways Will Push Today's AI To Become Prized AGIMay 05, 2025 am 11:08 AM
Big Bets On Which Of These Pathways Will Push Today's AI To Become Prized AGIMay 05, 2025 am 11:08 AMLet's explore the potential paths to Artificial General Intelligence (AGI). This analysis is part of my ongoing Forbes column on AI advancements, delving into the complexities of achieving AGI and Artificial Superintelligence (ASI). (See related art
 Do You Train Your Chatbot, Or Vice Versa?May 05, 2025 am 11:07 AM
Do You Train Your Chatbot, Or Vice Versa?May 05, 2025 am 11:07 AMHuman-computer interaction: a delicate dance of adaptation Interacting with an AI chatbot is like participating in a delicate dance of mutual influence. Your questions, responses, and preferences gradually shape the system to better meet your needs. Modern language models adapt to user preferences through explicit feedback mechanisms and implicit pattern recognition. They learn your communication style, remember your preferences, and gradually adjust their responses to fit your expectations. Yet, while we train our digital partners, something equally important is happening in the reverse direction. Our interactions with these systems are subtly reshaping our own communication patterns, thinking processes, and even expectations of interpersonal conversations. Our interactions with AI systems have begun to reshape our expectations of interpersonal interactions. We adapted to instant response,
 California Taps AI To Fast-Track Wildfire Recovery PermitsMay 04, 2025 am 11:10 AM
California Taps AI To Fast-Track Wildfire Recovery PermitsMay 04, 2025 am 11:10 AMAI Streamlines Wildfire Recovery Permitting Australian tech firm Archistar's AI software, utilizing machine learning and computer vision, automates the assessment of building plans for compliance with local regulations. This pre-validation significan
 What The US Can Learn From Estonia's AI-Powered Digital GovernmentMay 04, 2025 am 11:09 AM
What The US Can Learn From Estonia's AI-Powered Digital GovernmentMay 04, 2025 am 11:09 AMEstonia's Digital Government: A Model for the US? The US struggles with bureaucratic inefficiencies, but Estonia offers a compelling alternative. This small nation boasts a nearly 100% digitized, citizen-centric government powered by AI. This isn't
 Wedding Planning Via Generative AIMay 04, 2025 am 11:08 AM
Wedding Planning Via Generative AIMay 04, 2025 am 11:08 AMPlanning a wedding is a monumental task, often overwhelming even the most organized couples. This article, part of an ongoing Forbes series on AI's impact (see link here), explores how generative AI can revolutionize wedding planning. The Wedding Pl
 What Are Digital Defense AI Agents?May 04, 2025 am 11:07 AM
What Are Digital Defense AI Agents?May 04, 2025 am 11:07 AMBusinesses increasingly leverage AI agents for sales, while governments utilize them for various established tasks. However, consumer advocates highlight the need for individuals to possess their own AI agents as a defense against the often-targeted
 A Business Leader's Guide To Generative Engine Optimization (GEO)May 03, 2025 am 11:14 AM
A Business Leader's Guide To Generative Engine Optimization (GEO)May 03, 2025 am 11:14 AMGoogle is leading this shift. Its "AI Overviews" feature already serves more than one billion users, providing complete answers before anyone clicks a link.[^2] Other players are also gaining ground fast. ChatGPT, Microsoft Copilot, and Pe


Hot AI Tools

Undresser.AI Undress
AI-powered app for creating realistic nude photos

AI Clothes Remover
Online AI tool for removing clothes from photos.

Undress AI Tool
Undress images for free

Clothoff.io
AI clothes remover

Video Face Swap
Swap faces in any video effortlessly with our completely free AI face swap tool!

Hot Article
Hot Tools

SublimeText3 Linux new version
SublimeText3 Linux latest version

MinGW - Minimalist GNU for Windows
This project is in the process of being migrated to osdn.net/projects/mingw, you can continue to follow us there. MinGW: A native Windows port of the GNU Compiler Collection (GCC), freely distributable import libraries and header files for building native Windows applications; includes extensions to the MSVC runtime to support C99 functionality. All MinGW software can run on 64-bit Windows platforms.

SAP NetWeaver Server Adapter for Eclipse
Integrate Eclipse with SAP NetWeaver application server.

mPDF
mPDF is a PHP library that can generate PDF files from UTF-8 encoded HTML. The original author, Ian Back, wrote mPDF to output PDF files "on the fly" from his website and handle different languages. It is slower than original scripts like HTML2FPDF and produces larger files when using Unicode fonts, but supports CSS styles etc. and has a lot of enhancements. Supports almost all languages, including RTL (Arabic and Hebrew) and CJK (Chinese, Japanese and Korean). Supports nested block-level elements (such as P, DIV),

Dreamweaver CS6
Visual web development tools

