
Home >Technology peripherals >AI >The computational efficiency was increased by more than 100 times, and was submitted to Li Jinjin's team to develop a large model based on Transformer for ab initio molecular dynamics calculations.
The computational efficiency was increased by more than 100 times, and was submitted to Li Jinjin's team to develop a large model based on Transformer for ab initio molecular dynamics calculations.

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOriginal
- 2024-06-18 18:02:541236browse

Author | Tao Kehao
Accurate simulation of the dynamic behavior of atoms and molecules is crucial to the development of a new generation of high-efficiency materials important.
However, traditional ab initio molecular dynamics (AIMD) simulations naturally provide high-precision prediction capabilities, but their high computational cost and long simulation time greatly limit the progress of research.
For example, it usually takes several months to build a 30 picosecond simulation of a material system containing 100 atoms, which poses a huge challenge to the development of new materials that require rapid iteration and optimization.
In this context, an artificial intelligence model that can significantly speed up this process is of great value.
Faced with these challenges, the Shanghai Jiao Tong University Artificial Intelligence and Microstructure Laboratory (AIMS-lab) developed a revolutionary artificial intelligence model called T-AIMD.
This model adopts a Transformer-based network architecture, which can not only effectively reduce computational costs, but also quickly and accurately predict the behavior of any ion in any crystal structure.
In this way, the T-AIMD model speeds up traditional AIMD simulation by more than 100 times, significantly speeding up the material performance evaluation process.
In addition, the model successfully constructed a large database of mixed ion conductors and verified the accuracy of its predictions in multiple battery experiments.
This method has broad application potential not only in the fields of molecular dynamics modeling (MD), biopharmaceutical molecule binding targets, protein folding, material thermodynamic processes and mechanical property calculations.
also provides new methodologies for using generative artificial intelligence models to solve complex problems in a wider range of scientific fields.
The successful application of T-AIMD demonstrates the great potential of artificial intelligence technology in promoting scientific research and technological innovation, opening up new paths for future new material research and development and biological design development.
The research, titled "Transformer enables ion transport behavior evolution and conductivity regulation for solid electrolyte", was published in the internationally renowned journal "Energy Storage" on June 11, 2024 Materials》on.
The first author of the paper is Tao Kehao, a doctoral student in the Artificial Intelligence and Microstructure Laboratory of Shanghai Jiao Tong University, and the corresponding author is Professor Li Jinjin, director of the laboratory.

Article link: https://www.sciencedirect.com/science/article/pii/S2405829724003829
In artificial intelligence In the field, the Transformer model has become the preferred framework for processing complex sequence data due to its excellent parallel processing capabilities and excellent performance.
This model is particularly good at learning deep patterns and associations from large-scale data, so it has been widely used in language processing, image recognition, and various prediction tasks.
Despite this, the potential of Transformer has not yet been fully exploited in applications in materials science, especially in ab initio molecular dynamics (AIMD) simulations.
Traditional AIMD simulations are important in materials science for their ability to accurately simulate the dynamic behavior of atoms and molecules. However, such simulations often rely on repeated calculations and expensive experiments, which are not only time-consuming but also costly.
Faced with such challenges, an intelligent model that can quickly extract and process large amounts of sequence data is particularly important.
In response to this demand, the T-AIMD model developed by the AIMS-lab team of Shanghai Jiao Tong University uses the Transformer network architecture to significantly improve the speed and accuracy of AIMD simulation.
This new model can quickly and accurately analyze and predict the behavior of atoms and molecules under various conditions while greatly reducing computational costs.
Compared with traditional AIMD methods, T-AIMD can increase the simulation speed by more than 100 times, while maintaining high accuracy of prediction and significantly shortening the material development cycle.
This not only provides new tools for research in the field of materials science, but also demonstrates the application potential of AI in high-performance computing tasks, opening up new possibilities for future scientific exploration.
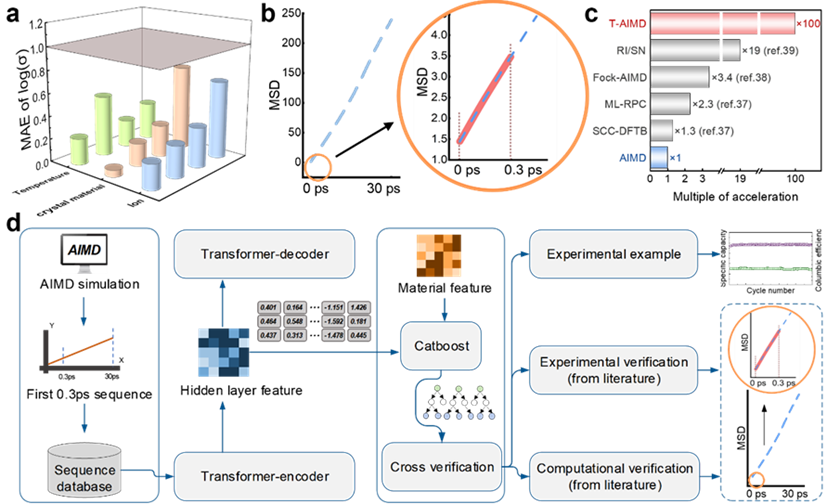
Illustration: T-AIMD prediction results and workflow diagram. (Source: paper)
Take solving the problem of predicting ion transport behavior in solid electrolytes as an example. By learning the diffusion sequence of ions in the electrolyte, the model is able to predict its behavior in future states, greatly accelerating the process of evaluating material properties.
In addition, the T-AIMD model also incorporates multi-source material descriptors, which enhances its application capabilities in processing complex material systems, allowing it to predict not only the behavior of a single ion species, but also multiple ions Interactions and complex dynamics problems in systems.
This new Transformer-based method provides a new perspective and tool for the development of solid electrolytes, and is expected to open up new research and application prospects in the field of materials science.
About how T-AIMD works
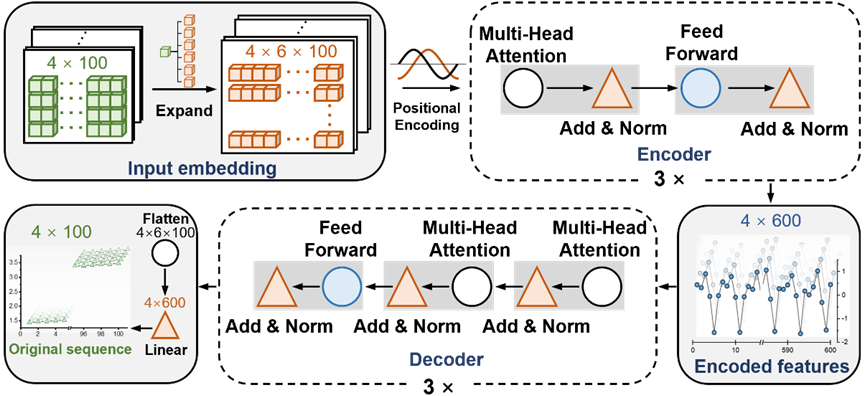
Illustration: T-AIMD’s network architecture diagram. (Source: Paper)
T-AIMD (Transformer-based Ab Initio Molecular Dynamics) is a model that combines ab initio molecular dynamics (AIMD) simulations and the Transformer deep learning architecture to improve solid-state electrolytes Speed and accuracy in predicting ion transport properties in materials. The working principle of this model can be divided into the following key steps:
1. Data preparation and preprocessing
T-AIMD first collects the ion diffusion data of the material , these data come from traditional AIMD simulations. The data generated by these simulations include time series data that record the movement of ions through the electrolyte. These sequence data are preprocessed and converted into a format suitable as input to the machine learning model.
2. Feature extraction
Using the encoder part of the Transformer model, T-AIMD can extract key features from sequence data. In this process, the model captures long-distance dependencies in the sequence through a self-attention mechanism, which is critical for understanding complex ion dynamics.
3. Sequence learning and prediction
After feature extraction, the decoder part of the Transformer model is used to perform sequence prediction based on the encoded features. In this step, the model can not only predict the future behavior of the ions, but also analyze the potential behavior of the ions under different conditions, such as different temperatures and pressures. In addition, the model can predict key performance indicators such as the ionic conductivity of the material through these learned features.
4. Integration of multi-source material descriptors
T-AIMD combines material descriptors from different sources, such as crystal structure, ion species and electronic properties, etc. , which helps the model more comprehensively understand and predict material properties. This integrated approach improves the model's versatility and adaptability in different material systems.
5. Model verification and application
The developed model needs to be compared with experimental data and other calculation methods to verify its prediction accuracy. After successful verification, T-AIMD can be used to quickly screen and optimize new target materials, greatly shortening the development cycle and reducing costs.
About the robust performance of T-AIMD
The robust performance of the T-AIMD model is mainly reflected in the following aspects:
1 , Accuracy
The T-AIMD model integrates the Transformer architecture, which greatly enhances its ability to learn and predict complex dynamic behaviors. In terms of AIMD simulation acceleration, T-AIMD shows higher accuracy than traditional methods. This is due to the application of deep learning technology, which enables the model to accurately predict ion behavior on longer time scales in a shorter time.
2. Computational efficiency
In terms of computational efficiency, T-AIMD is significantly better than the traditional AIMD method. Traditional AIMD simulations take a lot of time to simulate ion diffusion, but T-AIMD significantly reduces the dependence on high-performance computing resources by optimizing the calculation process, shortening the simulation time from months to days or hours.
3. Versatility and flexibility
T-AIMD can handle more complex data structures and Larger data sets. The model is able to adapt to many types of materials and effectively predict behavior under different environmental conditions, such as changes in temperature and pressure.
4. Model Robustness
T-AIMD shows high robustness when dealing with data with noise and uncertainty. In comparative experiments, T-AIMD can maintain a high degree of prediction accuracy even when the data is slightly biased, which is difficult to achieve by other simple machine learning models.
5. Scalability and adaptability
The architecture of the T-AIMD model allows flexible adjustment and optimization to adapt to changing research needs and new scientific discoveries. This scalability enables T-AIMD to continue to play a key role in future research, with applications beyond solid-state electrolytes extending to the study of other energy materials and complex chemical systems.
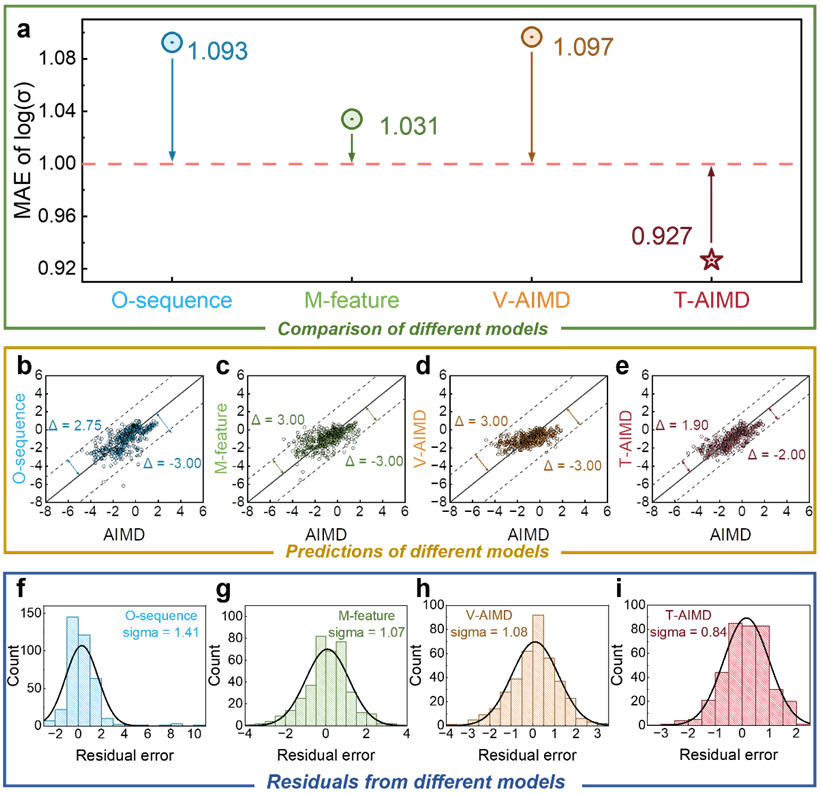
In summary, based on the T-AIMD framework, the simulation efficiency of molecular dynamics can be greatly accelerated, increasing the efficiency by 1,000 times, 10,000 times, or even more, providing opportunities for material manufacturing and biology. Design saves a lot of time and cost.
The T-AIMD model outperforms traditional AIMD simulations and other machine learning methods in several key aspects, and the examples given in the text show its strong potential and application prospects in solid-state electrolyte research and development.
T-AIMD’s utility goes far beyond that. The power and flexibility of this model make it widely applicable in many fields of materials science.
In the future, it is expected to be used to predict ion and molecular behavior in other types of materials such as semiconductors, metals and polymer materials.
In addition, the ability of the T-AIMD model is not limited to the prediction of the behavior of a single ion species. It can also handle complex interactions and dynamics problems in multi-ion systems, which makes it useful in designing new materials and improving existing materials. The performance of some materials has extremely high practical value.
The above is the detailed content of The computational efficiency was increased by more than 100 times, and was submitted to Li Jinjin's team to develop a large model based on Transformer for ab initio molecular dynamics calculations.. For more information, please follow other related articles on the PHP Chinese website!