
Heim >Technologie-Peripheriegeräte >KI >Vorhersage optimaler Optionen für die Synthese von Arzneimittelmolekülen mithilfe geometrischer Deep-Learning-Methoden und ebnet so den Weg für die Entdeckung neuer Arzneimittel
Vorhersage optimaler Optionen für die Synthese von Arzneimittelmolekülen mithilfe geometrischer Deep-Learning-Methoden und ebnet so den Weg für die Entdeckung neuer Arzneimittel

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBnach vorne
- 2024-01-13 22:36:071243Durchsuche

Postfunktionalisierung ist eine wirtschaftliche Methode zur Optimierung der Eigenschaften von Arzneimittelkandidaten. Allerdings macht die chemische Komplexität von Arzneimittelmolekülen die Funktionalisierung im Spätstadium oft zu einer Herausforderung.
Um dieses Problem zu lösen, haben Forscher der Universität München, der ETH Zürich und Roche Basel gemeinsam eine Plattform für die Funktionalisierung im Spätstadium entwickelt Deep Learning und Hochdurchsatz-Reaktionsscreening-Technologie
Angesichts der Tatsache, dass die Borylierung einer der Schlüsselschritte der Funktionalisierung ist, haben wir Computermodelle verwendet, um die Ausbeute unter verschiedenen Reaktionsbedingungen mit einem durchschnittlichen absoluten Fehlerbereich von 4–5 % vorherzusagen. Das Modell war in der Lage, neue Reaktionen für bekannte und unbekannte Substrate mit einer Genauigkeit von 92 % bzw. 67 % zu klassifizieren. Mit einem F-Score von 67 % für den Klassifikator konnten wir die Regioselektivität des Hauptprodukts genau erfassen. Bei der Anwendung auf 23 verschiedene kommerzielle Arzneimittelmoleküle haben wir erfolgreich viele Möglichkeiten zur strukturellen Diversifizierung entdeckt. Die Studie trägt den Titel „Using Geometric Deep Learning to Enable High-Throughput Experiments to Promote Late-stage Drug Diversification“ und wurde im November 2023 veröffentlicht in der Fachzeitschrift Nature Chemistry vom 23. März

Neuheit von Strukturen bei der Herstellung von Struktur-Aktivitäts-Beziehungen in der medizinischen Chemie und Komplexität machen die Synthese chemischer Zielstrukturen zu einer Herausforderung. Struktur-Aktivitäts-Beziehungsmodelle können Leitsubstanzen und Optimierungspläne für Leitsubstanzen leiten, um die pharmakologische Aktivität und die physikalisch-chemischen Eigenschaften von Arzneimittelkandidaten zu verbessern. Für die Erforschung von Struktur-Aktivitäts-Beziehungen ist eine effiziente Integration von entscheidender Bedeutung, die den Flaschenhals im Design-Herstellung-Test-Analyse-Zyklus darstellt
Es gibt viele alternative Methoden zur Aktivierung und Modifizierung von C-H-Bindungen für die Funktionalisierung organischer Gerüste im Spätstadium ( LSF), von molekularen Bausteinen bis hin zu fortschrittlichen pharmazeutischen Molekülen. Viele katalytische Systeme bieten gerichtete und ungerichtete Ansätze sowie einen chemischen und ortsselektiven Zugang zu modifizierten Analoga
Unter den zahlreichen LSF-Methoden gilt die C-H-Borylierungsmethode als die am häufigsten verwendete Methode zur schnellen Diversifizierung von Verbindungen. Organoborverbindungen können als zuverlässiges Mittel für nachfolgende C-C-Bindungskupplungsreaktionen in eine Vielzahl funktioneller Gruppen umgewandelt werden und ermöglichen so eine umfassende Erforschung der Struktur-Aktivitäts-Beziehungen.
Allerdings gibt es derzeit nur wenige Anwendungen von LSF in der Arzneimittelforschung. Die meisten dieser Berichte konzentrieren sich auf einen einzelnen LSF-Reaktionstyp. Die direkte LSF mehrerer Arten von C-H-Bindungen mit unterschiedlichen Bindungsstärken, elektronischen Eigenschaften sowie sterischen und funktionellen Gruppenumgebungen stellt Herausforderungen dar. Darüber hinaus ist die Durchführung von LSF-Projekten oft zeit- und ressourcenintensiv, was nicht mit den engen Zeitplänen und begrenzten Ressourcen vieler medizinischer Chemieprojekte vereinbar ist. Die Grafik zeigt einen Überblick über die Forschung zur Diversifizierung der Borisierung. (Datenquelle: Papier)
Künstliche Intelligenz unterstütztes LSF (Language Support Feature)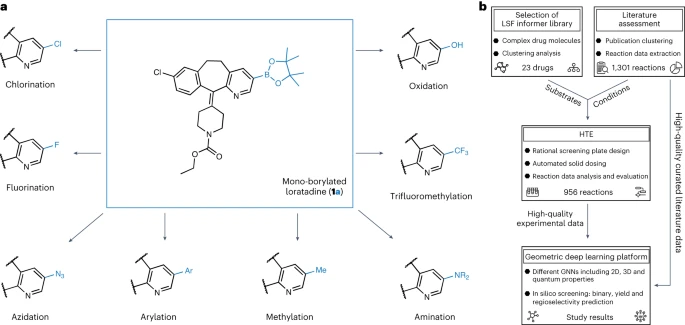
Allerdings sind diese Methoden auf kleine Molekülstrukturen und relativ kleine Datensätze beschränkt, was es schwierig macht Wenden Sie solche Modelle auf strukturell komplexere wirkstoffähnliche Moleküle an. Basierend auf Literaturrecherchen kann die Regioselektivität Iridium-katalysierter Borylierungsreaktionen durch ein hybrides maschinelles Lernmodell vorhergesagt werden, das mit quantenchemischen Informationen über Übergangszustände erweitert wird. Der Einfluss sterischer und elektronischer Effekte auf die Leistung von C-H-Aktivierungsreaktionsmodellen und ihre regioselektiven Anwendungen in Molekülen mit mehreren aromatischen Ringsystemen wurde jedoch noch nicht untersucht Automatisiertes LSF-Borisierungs-Screening mit geometrischem Deep Learning Forscher der Universität München, der ETH Zürich und Roche Pharmaceuticals Basel stellen eine automatisierte LSF-Borisierungs-Screening-Methode vor, die auf geometrisches Deep Learning angewendet wird, um Möglichkeiten zur Hit- und Lead-Diversifizierung im Spätstadium zu identifizieren. Computational Deep Learning wurde eingesetzt, um das Reaktionsergebnis, die Ausbeute und die Regioselektivität komplexer LSF-Wirkstoffmoleküle vorherzusagen. David Nippa, Hauptautor und Doktorand in der Forschungsgruppe an der Fakultät für Chemie und Pharmazie der LMU und Roche, sagte, dass mit diesem Ansatz die Anzahl der Laborexperimente deutlich reduziert und dadurch die Effizienz gesteigert werden könne chemische Synthese. und Nachhaltigkeit Für die ersten Schritte dieser Forschung führten wir eine gründliche Analyse der veröffentlichten Literatur durch, um geeignete Hochdurchsatz-Screening-Reaktionsbedingungen und Substrate auszuwählen, die für die Eigenschaften von Leitverbindungen im Spätstadium der Arzneimittelforschung relevant sind. Wir haben die Reaktionsbedingungen anhand eines manuell organisierten Literaturdatensatzes von 38 Artikeln ermittelt. Die Auswahl des LSF-Substrats basierte auf den Ergebnissen der Clusteranalyse von 1.174 zugelassenen Arzneimitteln, was zu 23 strukturell unterschiedlichen Arzneimittelmolekülen führte. Dieser Ansatz ermöglicht es Forschern, relevante Beispiele für Reaktionsbedingungen und Substrate in einem „Informationsbibliotheks“-Ansatz zu verwenden, anstatt sich ausschließlich auf ideale Substrate und Fragmente mit begrenzter Anwendbarkeit zu verlassen, um die Leitverbindungssynthese zu optimieren. Im zweiten Schritt verwenden Forscher semi -Automatisierte Hochdurchsatzexperimente (HTE) zur Generierung von Daten (experimentelle Datensätze). Die Reaktionsdaten der ausgewählten Arzneimittelmoleküle und Reaktionsbedingungen liefern hochwertige Daten für das maschinelle Lernen nachfolgender Reaktionsergebnisse. Schließlich trainierten wir verschiedene Graph-Neural-Network-Modelle (GNN) unter Verwendung zweidimensionaler, dreidimensionaler und atomarer Teilladungen Verstärkung Verwenden Sie molekulare Graphen, um binäre Reaktionsergebnisse, Reaktionsausbeuten und Regioselektivitäten vorherzusagen. „Interessanterweise verbesserten sich die Vorhersagen, wenn wir dreidimensionale Informationen über das Ausgangsmaterial und nicht nur seine zweidimensionale chemische Formel berücksichtigten“, sagte Kenneth Atz, Doktorand an der ETH Zürich. Dieser Ansatz wurde erfolgreich zur Identifizierung von Positionen eingesetzt innerhalb bestehender Wirkstoffe, wo zusätzliche reaktive Gruppen eingeführt werden können. Dies wird Forschern helfen, schneller neue, wirksamere Varianten bekannter pharmazeutischer Wirkstoffe zu entwickeln 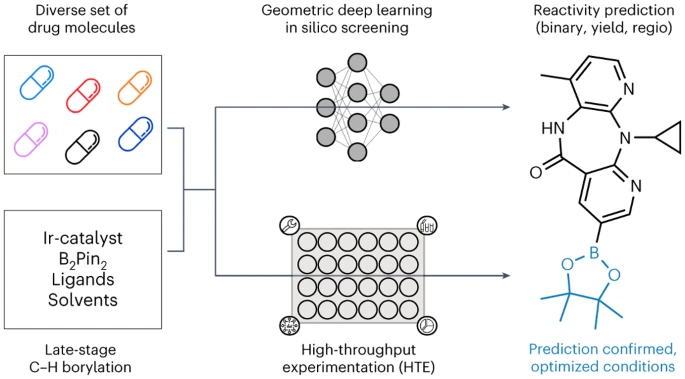
Das obige ist der detaillierte Inhalt vonVorhersage optimaler Optionen für die Synthese von Arzneimittelmolekülen mithilfe geometrischer Deep-Learning-Methoden und ebnet so den Weg für die Entdeckung neuer Arzneimittel. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Der Industriepark auf Dorfebene Foshan wird wiedergeboren: Die Auf- und Abstiegstreppen in der intelligenten Roboterfertigungsstadt sind die vor- und nachgelagerten, und der Industriepark ist die Industriekette
- Nutzen Sie Wettbewerbe zur Förderung von Lern- und Testergebnissen, um der Internet-of-Things-Branche zu einer qualitativ hochwertigen Entwicklung zu verhelfen
- Das erste Kollaborative Entwicklungsgipfelforum der Yangtze River Delta Robot Industry Chain 2023 wurde erfolgreich in Wuhu, Anhui, abgehalten
- Roboter-ETF (159770): Die „Leitmeinungen zur Innovation und Entwicklung humanoider Roboter' ziehen an vier aufeinanderfolgenden Tagen Nettokapitalzuflüsse an und können den industriellen Entwicklungsprozess fördern
- Roboterindustrie: das nächste heiße Feld im KI-Zeitalter, eine der neun großen Industrien der Zukunft!