
Heim >Technologie-Peripheriegeräte >KI >Die Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!
Die Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!

- WBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBOYWBnach vorne
- 2023-12-14 20:04:20669Durchsuche
Chemische Reaktionen sind die Grundlage des Arzneimitteldesigns und der Forschung in der organischen Chemie. In der Forschungsgemeinschaft besteht ein wachsender Bedarf an einem groß angelegten Deep-Learning-Framework, das die grundlegenden Regeln chemischer Reaktionen effektiv erfassen kann.
Kürzlich hat ein Forschungsteam der Peking-Universität und Wangshi Intelligence eine neue Methode vorgeschlagen, um die Lücke zwischen reaktionsbasiertem molekularem Vortraining und Generierungsaufgaben zu schließen.
Inspiriert von den Mechanismen der organischen Chemie haben Forscher ein neues Pre-Training-Framework entwickelt, das es ermöglicht, induktive Voreingenommenheit in Modelle zu integrieren. Dieses vorgeschlagene Framework erzielt Ergebnisse auf dem neuesten Stand der Technik bei der Durchführung anspruchsvoller nachgelagerter Aufgaben. Durch die Nutzung chemischer Kenntnisse überwindet das Framework die Einschränkungen aktueller Modelle zur Molekülerzeugung, die auf einer kleinen Anzahl von Reaktionsvorlagen basieren. In umfangreichen Experimenten generierte das Modell hochwertige, synthetisierbare wirkstoffähnliche Strukturen
Insgesamt ist diese Forschung ein wichtiger Schritt hin zu einem groß angelegten Deep-Learning-Framework für eine Vielzahl reaktionsbasierter Anwendungen.
Die Studie trug den Titel „Überbrückung der Lücke zwischen chemischem Reaktions-Pretraining und bedingter Molekülgenerierung mit einem einheitlichen Modell“ und wurde am 5. Dezember 2023 in „Nature Machine Intelligence“ veröffentlicht.

Link zum Papier: https://www.nature.com/articles/s42256-023-00764-9
Deep-Learning-Modelle werden in vielen wissenschaftlichen Forschungsbereichen häufig eingesetzt. Das Pre-Training-Framework spielt eine positive Rolle bei der nahtlosen Integration neuer Aufgaben und kann den Modellierungsprozess beschleunigen, insbesondere wenn die markierten Daten begrenzt sind.
Die Grundlage des Arzneimitteldesigns und der Forschung in der organischen Chemie sind chemische Reaktionen. Derzeit haben die Forschung und Anwendung von Data Mining den Einsatz von Deep-Learning-Modellen in chemischen Reaktionen ermöglicht. Basierend auf diesen Daten gab es viele datengesteuerte Studien, die sich mit dem Repräsentationslernen chemischer Reaktionen befassten.
Repräsentationslernen bezieht sich auf das automatische Lernen nützlicher Funktionen aus Daten und deren anschließende Verwendung für verschiedene nachgelagerte Aufgaben. Bestehende Methoden ignorieren grundlegende Theorien der organischen Chemie und schränken ihre Leistung ein.
Molekülgenerierung auf Basis chemischer Reaktionen
Neben der Reaktionsklassifizierungsaufgabe ist auch die Molekülgenerierung auf Basis chemischer Reaktionen eine wichtige Anwendung. In frühen Studien wurden häufig templatbasierte Strategien zur schrittweisen Molekülgenerierung übernommen
Diese templatbasierten Methoden stützten sich stark auf vordefinierte Bausteine und Reaktionen, was den zugänglichen chemischen Raum einschränkte. Ein ähnlicher Trend ist im Bereich der Reaktionsproduktvorhersage zu beobachten, wo templatbasierte Methoden nicht auf komplexe Reaktionen übertragen werden können; dieses Problem kann durch den Einsatz templatfreier Methoden gelöst werden.
Bei reaktionsbasierten Molekülgenerierungsaufgaben zeigen templatfreie Methoden auch Generalisierungsvorteile gegenüber templatbasierten Methoden. Allerdings können bestehende templatfreie Methoden zur Molekülgenerierung nur Moleküle erzeugen, die auf vordefinierten Reaktantenbibliotheken basieren. Darüber hinaus ist es für die Leitverbindungs- oder Leitstrukturoptimierungsphase im Arzneimitteldesign vorteilhafter, chemische Reaktionen als Bearbeitungswerkzeuge zu nutzen, um eine bestimmte Struktur zu modifizieren. Die resultierende chemische Bibliothek wird sich auf einen Teilbereich des chemischen Raums konzentrieren, der mit weniger Reaktionsschritten synthetisiert werden kann.
Ein neues, umfassendes Deep-Learning-Framework für chemische Reaktionen
Hier schlagen Forscher ein neues, umfassendes Deep-Learning-Framework für chemische Reaktionen vor, genannt Uni-RXN. Ziel ist es, zwei grundlegende Aufgaben zu lösen: selbstüberwachtes Repräsentationslernen und bedingte generative Modellierung.
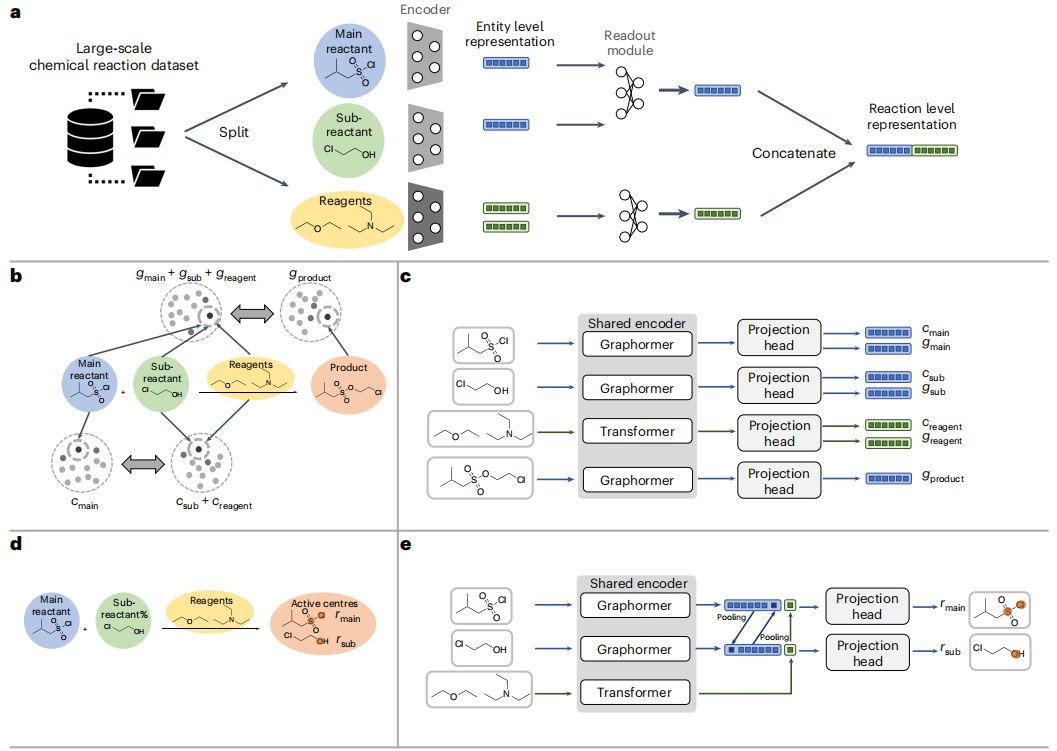
Anzeigen: Zusammensetzung und Methode von Uni-RXN. (Zitiert aus: Artikel)
Anders als bestehende Methoden schlugen die Forscher eine Reihe selbstüberwachter Aufgaben vor, die speziell für chemische Reaktionen entwickelt wurden. Zu diesen Aufgaben gehören die Vorhersage des Reaktionszentrums, die Paarung von Primärreaktanten und Unterreaktanten sowie die Paarung von Reaktanten und Produkten. In einer umfassenden Bewertung anspruchsvoller Reaktionsaufgaben übertrifft die Uni-RXN-Methode den Stand der Technik und demonstriert ihre Fähigkeit, Domänenwissen über chemische Reaktionen effektiv zu erfassen. Die erzielten vielversprechenden Ergebnisse ebnen den Weg für weitreichende nachgelagerte Anwendungen
Durch die effektive Erfassung chemischer Regeln ist Uni-RXN ideal für Erzeugungsaufgaben geeignet. Im Gegensatz zu herkömmlichen Methoden, die auf der Auswahl von Fragmenten aus einer Bibliothek vordefinierter Reaktanten basieren, verwendet Uni-RXN molekulare Strukturen als Eingabebedingungen und generiert Darstellungen der entsprechenden Reaktanten, während die Permutationsinvarianz innerhalb der Reaktion erhalten bleibt. Uni-RXN nutzt die Leistungsfähigkeit des Suchpakets für dichte Vektorähnlichkeiten und ermöglicht die effiziente Suche nach Reaktanten aus großen Reaktanten- und Reagenzienbibliotheken. Anschließend wird ein Reaktionsvorhersagemodell verwendet, um Produktergebnisse zu generieren.
Im Vergleich zu templatbasierten Methoden, die nur einen begrenzten Teilbereich des chemischen Raums erforschen, weist Uni-RXN eine überlegene Leistung bei der Erzeugung eines breiteren Spektrums synthetisierbarer arzneimittelähnlicher Strukturen auf. Aufgrund dieser Funktion eignet es sich besonders für die Aufzählung virtueller Bibliotheken und wird durch umfassende statistische Analysen und Fallstudien unterstützt.
Die Uni-RXN-Methode bietet viele Vorteile und kann umfangreiche Darstellungen für die anspruchsvolle Aufgabe der Klassifizierung chemischer Reaktionen generieren. Im Vergleich zu anderen Basismodellen erreicht Uni-RXN eine Genauigkeit von 58,7 % mit nur 4 Datenpunkten pro Klasse. Umgeschriebener Inhalt: Die Genauigkeit der Klassifizierung chemischer Reaktionen ist in Tabelle 1 dargestellt. (Quelle: Paper)
Transformer-Modell kann verwendet werden, um zwischen optimierten und nicht optimierten chemischen Reaktionsdaten zu unterscheiden. Darüber hinaus kann der Encoder auch problemlos auf die Generierung struktureller Bedingungen angewendet werden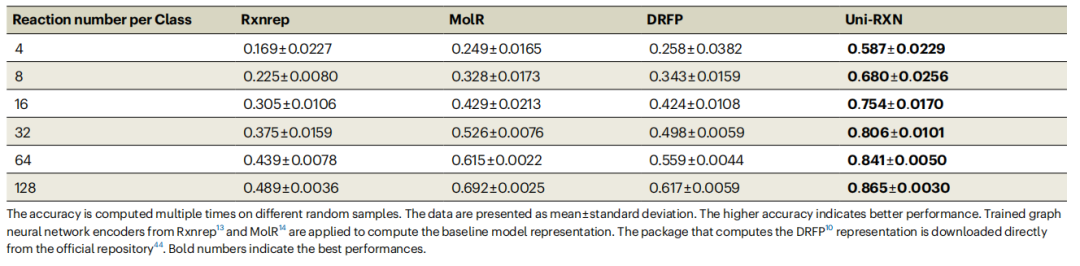
Der Inhalt, der neu geschrieben werden muss, ist: Das Diagramm zeigt die Abrufleistung und das Aufmerksamkeitsgewicht von Uni-RXN. (Quelle: Papier) 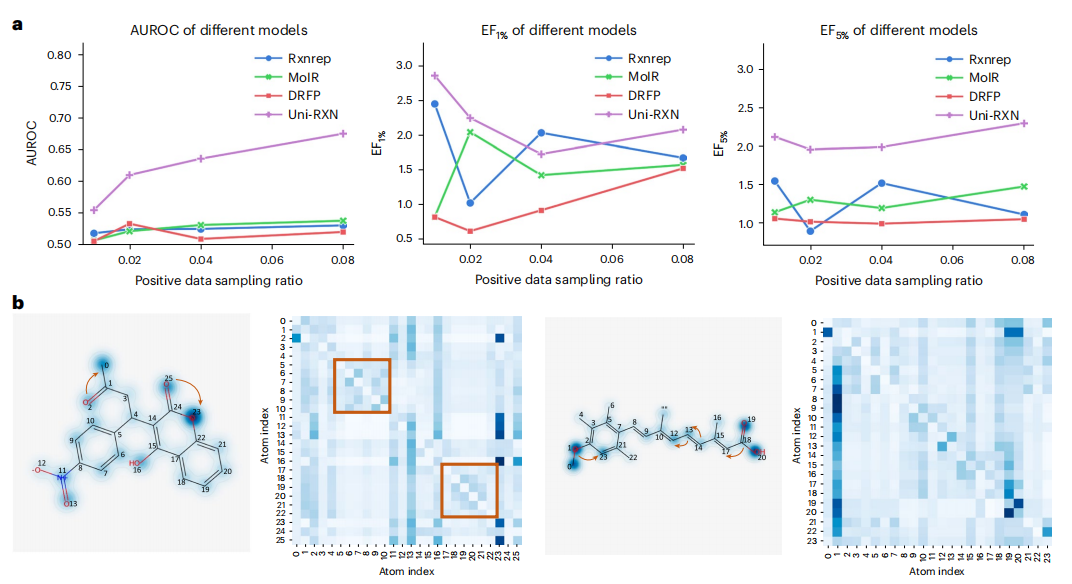
Die Ergebnisse verdeutlichen die günstigen Eigenschaften der durch das vorgeschlagene Modell erzeugten Moleküle, die sie für Aufgaben der Arzneimittelforschung gut geeignet machen. Dieses Modell kann mehr Moleküle mit arzneimittelähnlichen Eigenschaften und Synthetisierbarkeit erzeugen
Abbildung: Uni-RXNGen-Prozess und Leistung. (Quelle: Papier) 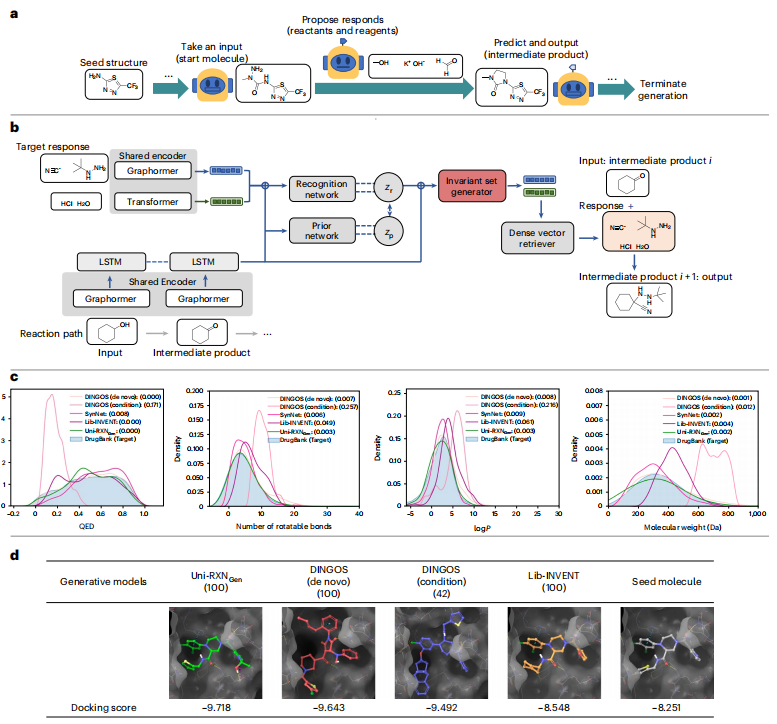
In Kombination mit virtuellen Screening-Methoden wie Molecular Docking kann dieses generierte Modell eine effiziente Struktur-Aktivitäts-Beziehungsforschung ermöglichen. Der riesige, durch dieses Modell erzeugte Raum für synthetische drogenähnliche Chemikalien kann die tatsächliche positive Rate bei der Wiederverwendung von Medikamenten oder bei der Suche nach Treffermolekülen verbessern.
Das obige ist der detaillierte Inhalt vonDie Peking-Universität und Wangshi Intelligence schlagen ein neues Modell vor: die Lücke zwischen dem Vortraining chemischer Reaktionen und der bedingten Molekülgenerierung zu schließen!. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Welche drei Arten von Datenbankdatenmodellen gibt es?
- Wie läuft beim Datenbankdesign die Konvertierung eines ER-Diagramms in ein relationales Datenmodell ab?
- Was sind die gängigen Softwareentwicklungsmodelle?
- Auf welcher Ebene des osi-Modells ist die Pfadauswahlfunktion abgeschlossen?
- Die All-in-One-Maschine H1 Neo zum Waschen und Trocknen von Steinmolekularsieben steht kurz vor der Veröffentlichung und die „dritte Trocknungstechnologie' ist zum größten Highlight geworden