
Heim >Technologie-Peripheriegeräte >KI >Implementierung einer ladungsbasierten Atomsimulation unter Verwendung des vorab trainierten universellen neuronalen Netzwerks CHGNet
Implementierung einer ladungsbasierten Atomsimulation unter Verwendung des vorab trainierten universellen neuronalen Netzwerks CHGNet

- PHPznach vorne
- 2023-09-27 17:45:01724Durchsuche
Umgeschrieben als: Violet
Groß angelegte Simulationen komplexer Elektronenwechselwirkungen bleiben eine der größten Herausforderungen in der Atommodellierung. Während klassische Kraftfelder oft nicht in der Lage sind, Kopplungen zwischen elektronischen Zuständen und Ionenumlagerungen zu beschreiben, leiden genauere Ab-initio-Moleküldynamiken unter der Rechenkomplexität, die lange und groß angelegte Simulationen verhindert, die für Forschungstechniken relevant sind. Das Phänomen ist von entscheidender Bedeutung
Kürzlich Forscher von der University of California, Berkeley, und dem Lawrence Berkeley National Laboratory schlugen ein MLIP-Modell (Machine Learning Interatomic Potential) vor, das auf graphischen neuronalen Netzen basiert: Crystal Hamiltonian Graph Neural Network (CHGNet), das die universelle potentielle Energie modellieren kann Oberflächen.
Die Forschung unterstreicht die Bedeutung von Ladungsinformationen für die Erfassung geeigneter chemischer Reaktionen und liefert Einblicke in ionische Systeme mit zusätzlichen elektronischen Freiheitsgraden, die mit früheren MLIPs nicht beobachtet werden konnten.
Die Forschung trug den Titel „CHGNet als vortrainiertes universelles neuronales Netzwerkpotenzial für ladungsinformierte atomistische Modellierung“ und wurde am 14. September 2023 in „Nature Machine Intelligence“ veröffentlicht.

Großmaßstäbliche Simulationen wie die Molekulardynamik (MD) sind wichtige Werkzeuge für die rechnerische Erforschung von Festkörpermaterialien. Die genaue Modellierung elektronischer Wechselwirkungen und ihrer subtilen Auswirkungen in Molekulardynamiksimulationen bleibt jedoch eine große Herausforderung. Empirische Methoden wie klassische Kraftfelder sind oft nicht genau genug, um komplexe elektronische Wechselwirkungen zu erfassen.
Ab-initio-Molekulardynamik (AIMD) in Kombination mit Dichtefunktionaltheorie (DFT) kann verwendet werden, um Elektronen innerhalb der Dichtefunktionalnäherungsstruktur explizit zu berechnen und so hohe Ergebnisse zu erzielen -treue Ergebnisse mit quantenmechanischer Präzision. Langfristige, groß angelegte spinpolarisierte AIMD-Simulationen, die für die Untersuchung von Ionenmigration, Phasenübergängen und chemischen Reaktionen von entscheidender Bedeutung sind, sind anspruchsvoll und rechenintensiv.
MLIPs wie ænet und DeepMD bieten vielversprechende Lösungen, um die Lücke zwischen teuren elektronischen Strukturmethoden und effizienten klassischen interatomaren Potentialen zu schließen. Die Einbeziehung der wichtigen Auswirkungen der Valenz auf die chemische Bindung bleibt jedoch eine Herausforderung in MLIP.
Ladung kann auf viele Arten dargestellt werden, von einfachen Oxidationszustandsbezeichnungen bis hin zu kontinuierlichen Wellenfunktionen, die aus der Quantenmechanik abgeleitet sind. Die Herausforderung bei der Integration von Gebühreninformationen in MLIP ergibt sich aus vielen Faktoren, wie etwa der Mehrdeutigkeit der Darstellung, der Komplexität der Interpretation, dem Mangel an Etiketten usw.
Was neu geschrieben werden muss, ist: CHGNet-Architektur
CHGNet basierend auf dem Materialprojekt Der Flugbahndatensatz (MPtrj) ist auf Energie, Kraft, Spannung und magnetisches Moment vorab trainiert. Der Datensatz enthält mehr als 10 Jahre Dichtefunktionaltheorie-Berechnungen für 1,5 Millionen anorganische Strukturen. Durch die explizite Einbeziehung magnetischer Momente ist CHGNet in der Lage, die Orbitalbelegung von Elektronen zu lernen und genau darzustellen, wodurch seine Fähigkeit zur Beschreibung atomarer und elektronischer Freiheitsgrade verbessert wird. Inhalt schreiben: Abbildung: Die Verteilung von Elementen im MPtrj-Datensatz. (Quelle: Paper) Hier definieren Forscher Ladung als eine atomare Eigenschaft (atomare Ladung), die durch Einbeziehung magnetischer Momente (Magmoms) abgeleitet werden kann. Untersuchungen zeigen, dass durch die explizite Einbindung ortsspezifischer Magmoms als Ladungszustandsbeschränkungen in CHGNet die Latentraum-Regularisierung verbessert und elektronische Wechselwirkungen genau erfasst werden können.
Die Grundlage von CHGNet ist GNN, bei dem Graphfaltungsschichten verwendet werden. Atomare Informationen werden durch sie verbreitet eine Menge von Knoten {vi}, die durch Kanten {eij} verbunden sind. Translations-, Rotations- und Ausrichtungsinvarianz bleiben in GNN erhalten. CHGNet nimmt als Eingabe eine Kristallstruktur mit unbekannten Atomladungen und gibt die entsprechenden Energien, Kräfte, Spannungen und Magmoms aus. Ladungsdekorierte Strukturen können aus Feldmagmomen und der Atomorbitaltheorie abgeleitet werden.

Der umgeschriebene Inhalt lautet wie folgt: Abbildung: CHGNet-Modellarchitektur. (Quelle: Papier)
In CHGNet werden periodische Kristallstrukturen in Atomgraphen umgewandelt  , indem nach benachbarten Atomen vj innerhalb
, indem nach benachbarten Atomen vj innerhalb  jedes Atoms vi in der Originaleinheit gesucht wird.
jedes Atoms vi in der Originaleinheit gesucht wird.
Im Gegensatz zu anderen GNNs, bei denen aktualisierte Atommerkmale 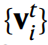 nach t Faltungsschichten direkt zur Energievorhersage verwendet werden, reguliert CHGNet Knotenmerkmale
nach t Faltungsschichten direkt zur Energievorhersage verwendet werden, reguliert CHGNet Knotenmerkmale
 nach t−1 Faltungsschichten, um Informationen über das Magma einzubeziehen. Regularisierte Merkmale
nach t−1 Faltungsschichten, um Informationen über das Magma einzubeziehen. Regularisierte Merkmale  enthalten umfassende Informationen über die lokale Ionenumgebung und Ladungsverteilung. Daher ist die atomare Signatur
enthalten umfassende Informationen über die lokale Ionenumgebung und Ladungsverteilung. Daher ist die atomare Signatur  , die zur Vorhersage von Energie, Kraft und Spannung verwendet wird, die Ladung, die durch Informationen über ihren Ladungszustand eingeschränkt wird. Daher kann CHGNet Informationen zum Ladungszustand bereitstellen, indem es nur Kernpositionen und Atomidentitäten als Eingaben verwendet, was die Untersuchung der Ladungsverteilung in der Atommodellierung ermöglicht.
, die zur Vorhersage von Energie, Kraft und Spannung verwendet wird, die Ladung, die durch Informationen über ihren Ladungszustand eingeschränkt wird. Daher kann CHGNet Informationen zum Ladungszustand bereitstellen, indem es nur Kernpositionen und Atomidentitäten als Eingaben verwendet, was die Untersuchung der Ladungsverteilung in der Atommodellierung ermöglicht.
Anwendungen von CHGNet in Festkörpermaterialien
Forscher demonstrierten mehrere Anwendungen von CHGNet in Festkörpermaterialien. Demonstriert Ladungsbeschränkungen und Latentraumregularisierung atomarer Ladungen in Na2V2(PO4)3 und demonstriert den Ladungstransfer und Phasenübergang von CHGNet in LixMnO2, die Elektronenentropie im LixFePO4-Phasendiagramm und den granatartigen Lithium-Superionenleiter Li3+ Lithium (Li) und Diffusionsfähigkeit in xLa3Te2O12.
Um die Behandlung atomarer Ladungen zu rationalisieren, wird das Natriumionen-Kathodenmaterial Na4V2(PO4)3 vom NASICON-Typ als anschauliches Beispiel verwendet. Zusätzlich zum Lernen aus der räumlichen Koordination von V-Kernen unterscheidet CHGNet erfolgreich V-Ionen in zwei Gruppen, dreiwertiges V und vierwertiges V, ohne vorherige Kenntnisse über die Ladungsverteilung von V-Ionen.

Die Abbildung zeigt die Normalisierung des magnetischen Moments und des verborgenen Raums in Na2V2(PO4)3. (Zitiert aus dem Artikel)
Die Untersuchung von LixFePO4 hat die Fähigkeit von CHGNet hervorgehoben,

zu unterscheiden, was für die Einbeziehung von Elektronenentropie und endlicher Temperaturphasenstabilität von entscheidender Bedeutung ist.

Abbildung: LixFePO4-Phasendiagramm von CHGNet. (Quelle: Papier)
In der Untersuchung von LiMnO2 wurde gezeigt, dass CHGNet durch langfristige Ladungsinformation MD Einblicke in den Zusammenhang zwischen Ladungsdisproportionierung und Phasenübergang in heterovalenten Übergangsmetalloxidsystemen gewinnen kann.
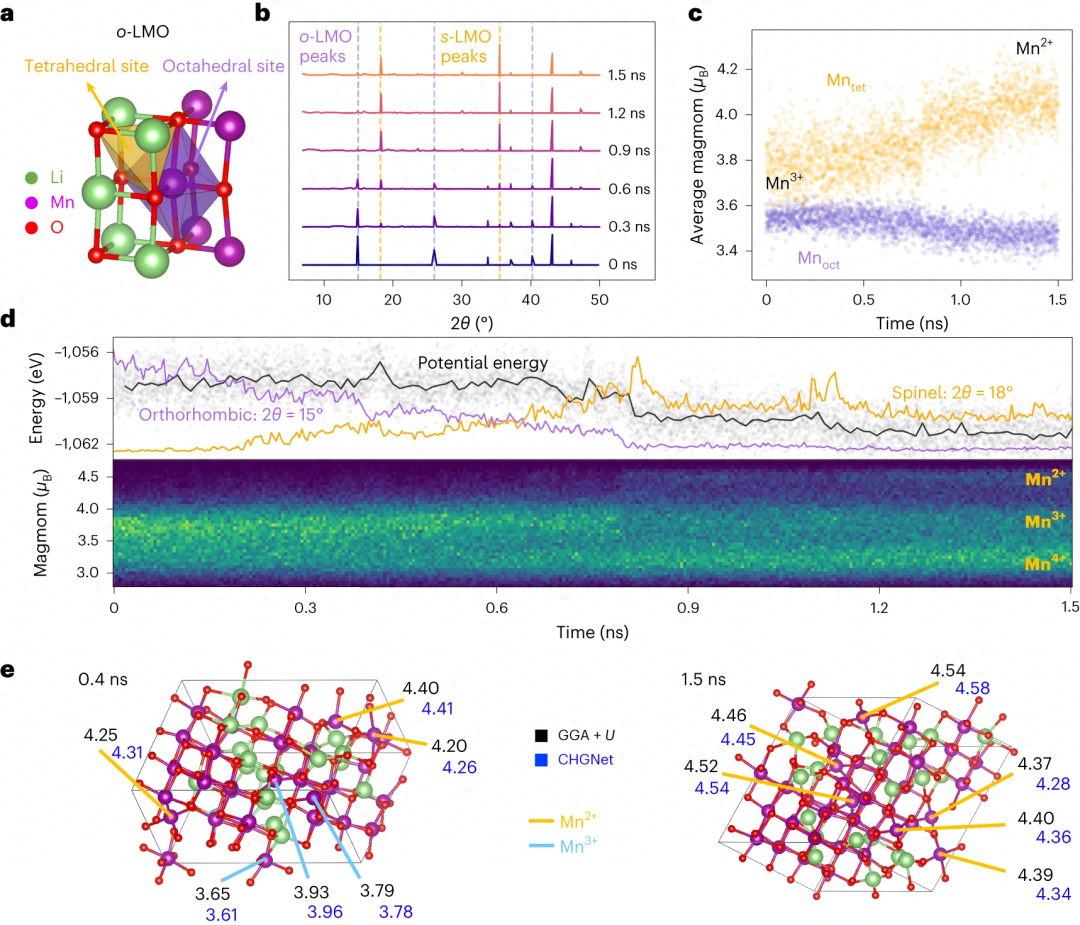
Umgeschriebener Inhalt: Abbildung: Phasenübergang und Ladungsdifferenzierung von Li0,5MnO2. (Zitat aus: Papier)
Als nächstes untersuchten wir die Genauigkeit von CHGNet in allgemeinen Molekulardynamiksimulationen. Wir untersuchen die Lithiumdiffusion in Granatleitern

Abbildung: Lithiumdiffusionsfähigkeit in Granat Li3La3Te2O12. (Quelle: Papier)
Die Ergebnisse zeigen, dass CHGNet nicht nur den Aktivierungs-Diffusions-Netzwerkeffekt genau erfassen kann, sondern auch seine Aktivierungsenergie sehr gut mit den DFT-Ergebnissen übereinstimmt. Dies beweist, dass CHGNet die starke Wechselwirkung von Lithiumionen in der lokalen Umgebung genau erfassen kann und in der Lage ist, ein stark nichtlineares Diffusionsverhalten zu simulieren. Darüber hinaus ist CHGNet in der Lage, den Fehler bei simulierten Diffusionsraten deutlich zu reduzieren, und durch die Ausweitung auf Nanosekundensimulationen ist es möglich, Systeme mit schlechten Diffusionsraten zu untersuchen
Kann weiter verbessert werden
Obwohl die oben genannten Fortschritte erzielt wurden Es gibt noch Raum für weitere Verbesserungen im Weltraum
Erstens gewährleistet die Verwendung von Magmom für die Valenzinferenz nicht unbedingt die globale Ladungsneutralität
Zweitens ist Magmom zwar eine gute Heuristik für die Berechnung der Atomladung für die Spinpolarisation in ionischen Systemen, es ist jedoch klar, dass die Schlussfolgerung der Atomladung für nichtmagnetische Ionen mehrdeutig sein kann, sodass zusätzliche Domänenkenntnisse erforderlich sind. Daher kann bei Ionen ohne Magmom das atomzentrierte Magmom seine Atomladung nicht genau wiedergeben. CHGNet leitet die Ladung aus der Umgebung ab, ähnlich wie die Funktionalität anderer MLIPs. z.B. Elektronenpositionierungsfunktionen, elektrische Polarisation und Partitionierung basierend auf Atomorbitalen. Diese Methoden können für die Entwicklung atomarer Merkmale im latenten Raum verwendet werden.
CHGNet ermöglicht ladungsbasierte Atomsimulationen, die sich für groß angelegte Computersimulationen zur Untersuchung heterovalenter Systeme eignen, und erweitert so den Umfang der Forschungsmöglichkeiten in den Bereichen Computerchemie, Physik, Biologie und Materialwissenschaften über Ladungsübertragungskopplungsphänomene
Bitte klicken Sie auf den folgenden Link, um das Papier anzuzeigen: https://www.nature.com/articles/s42256-023-00716-3
Das obige ist der detaillierte Inhalt vonImplementierung einer ladungsbasierten Atomsimulation unter Verwendung des vorab trainierten universellen neuronalen Netzwerks CHGNet. Für weitere Informationen folgen Sie bitte anderen verwandten Artikeln auf der PHP chinesischen Website!
In Verbindung stehende Artikel
Mehr sehen- Grundlegende Java-Theorie und Klassifizierung der Programmiersprache
- Vorgestellt in der internationalen Spitzenpublikation PNAS! Ausgehend von theoretischen Computern schlugen Wissenschaftler ein Bewusstseinsmodell vor – eine „bewusste Turing-Maschine'.
- Theorie, Implementierung und Hyperparameter-Tuning von Entscheidungsbäumen und Zufallswäldern